Zyvox 600mg Linezolid Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce que Zyvox 600 mg et comment est-il utilisé ?
Zyvox est un médicament délivré sur ordonnance utilisé pour traiter les symptômes d'infections bactériennes telles que la pneumonie, les infections cutanées et les infections résistantes à d'autres antibiotiques. Zyvox 600 mg peut être utilisé seul ou avec d'autres médicaments.
Zyvox appartient à une classe de médicaments appelés antibiotiques, autres ; Oxazolidinones.
On ne sait pas si Zyvox est sûr et efficace chez les enfants.
Quels sont les effets secondaires possibles du Zyvox ?
Zyvox peut provoquer des effets secondaires graves, notamment :
- urticaire,
- difficulté à respirer,
- gonflement du visage ou de la gorge,
- réaction cutanée sévère,
- fièvre,
- mal de gorge,
- Yeux brûlants,
- douleurs cutanées,
- éruption cutanée rouge ou violette avec cloques et desquamation,
- problèmes de vue,
- changements dans votre vision,
- fortes douleurs à l'estomac,
- diarrhée aqueuse ou sanglante,
- convulsions,
- transpiration,
- se sentir anxieux ou tremblant,
- agitation,
- hallucinations,
- fièvre,
- frissons,
- rythme cardiaque rapide,
- rigidité musculaire,
- contractions musculaires,
- perte de coordination,
- nausée,
- vomissement,
- diarrhée,
- douleurs musculaires inhabituelles,
- difficulté à respirer,
- Douleur d'estomac,
- rythme cardiaque irrégulier,
- vertiges,
- avoir froid,
- se sentir très faible ou fatigué,
- confusion,
- plaies buccales,
- plaies cutanées,
- ecchymoses faciles,
- saignement inhabituel,
- peau pâle,
- mains et pieds froids,
- étourdissements, et
- essoufflement
Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires les plus courants de Zyvox incluent :
- nausée,
- vomissement,
- diarrhée,
- légère éruption cutanée,
- anémie (faible nombre de globules rouges),
- maux de tête, et
- vertiges
Dites au médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles du Zyvox. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
LA DESCRIPTION
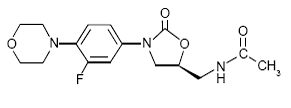
ZYVOX 600mg IV Injection, les comprimés ZYVOX et ZYVOX pour suspension buvable contiennent du linézolide, qui est un agent antibactérien synthétique de la classe des oxazolidinones. Le nom chimique du linézolide est (S)-N-[[3-[3-Fluoro-4-(4morpholinyl)phényl]-2-oxo-5-oxazolidinyl]méthyl]-acétamide.
La formule empirique est C16H20FN3O4. Son poids moléculaire est de 337,35 et sa structure chimique est représentée ci-dessous :
ZYVOX 600mg IV Injection est fourni sous forme de solution isotonique stérile prête à l'emploi pour perfusion intraveineuse. Chaque mL contient 2 mg de linézolide. Les ingrédients inactifs sont le citrate de sodium, l'acide citrique et le dextrose dans un véhicule aqueux pour administration intraveineuse. La teneur en sodium (Na+) est de 0,38 mg/mL (5 mEq/poche de 300 mL et 1,7 mEq/poche de 100 mL).
Le comprimé ZYVOX pour administration orale contient 600 mg de linézolide sous forme de comprimé pelliculé. Les ingrédients inactifs sont l'amidon de maïs, la cellulose microcristalline, l'hydroxypropylcellulose, le glycolate d'amidon sodique, le stéarate de magnésium, l'hypromellose, le polyéthylène glycol, le dioxyde de titane et la cire de carnauba. La teneur en sodium (Na+) est de 2,92 mg par comprimé de 600 mg (0,1 mEq/comprimé).
ZYVOX 600 mg pour suspension buvable est fourni sous forme de granule/poudre aromatisé à l'orange pour constitution en une suspension pour administration orale. Après constitution, chaque 5 mL contient 100 mg de linézolide. Les ingrédients inactifs sont le saccharose, l'acide citrique, le citrate de sodium, la cellulose microcristalline et la carboxyméthylcellulose sodique, l'aspartame, la gomme xanthane, le mannitol, le benzoate de sodium, le dioxyde de silicium colloïdal, le chlorure de sodium et les arômes [voir INFORMATIONS PATIENTS ]. La teneur en sodium (Na+) est de 8,52 mg/5 mL (0,4 mEq/5 mL).
LES INDICATIONS
Pneumonie nosocomiale
ZYVOX est indiqué pour le traitement de la pneumonie nosocomiale causée par Staphylococcus aureus (isolats sensibles et résistants à la méthicilline) ou Streptococcus pneumoniae [voir Etudes cliniques ].
Pneumonie communautaire
ZYVOX 600 mg est indiqué pour le traitement de la pneumonie communautaire causée par Streptococcus pneumoniae, y compris les cas avec bactériémie concomitante, ou Staphylococcus aureus (isolats sensibles à la méthicilline uniquement) [voir Etudes cliniques ].
Infections compliquées de la peau et des structures cutanées
ZYVOX 600 mg est indiqué pour le traitement des infections compliquées de la peau et des annexes cutanées, y compris les infections du pied diabétique, sans ostéomyélite concomitante, causées par Staphylococcus aureus (isolats sensibles et résistants à la méthicilline), Streptococcus pyogenes ou Streptococcus agalactiae. ZYVOX n'a pas été étudié dans le traitement des ulcères de décubitus [voir Etudes cliniques ].
Infections non compliquées de la peau et des structures cutanées
ZYVOX 600 mg est indiqué pour le traitement des infections non compliquées de la peau et des tissus cutanés causées par Staphylococcus aureus (isolats sensibles à la méthicilline uniquement) ou Streptococcus pyogenes [voir Etudes cliniques ].
Infections à Enterococcus faecium résistant à la vancomycine
ZYVOX 600 mg est indiqué pour le traitement des infections à Enterococcus faecium résistant à la vancomycine, y compris les cas avec bactériémie concomitante [voir Etudes cliniques ].
Limites d'utilisation
- ZYVOX 600 mg n'est pas indiqué dans le traitement des infections à Gram négatif. Il est essentiel qu'un traitement Gram négatif spécifique soit initié immédiatement si un agent pathogène Gram négatif concomitant est documenté ou suspecté [voir AVERTISSEMENTS ET PRECAUTIONS ].
- L'innocuité et l'efficacité des formulations de ZYVOX 600 mg administrées pendant plus de 28 jours n'ont pas été évaluées dans des essais cliniques contrôlés [voir Etudes cliniques ].
Usage
Pour réduire le développement de bactéries résistantes aux médicaments et maintenir l'efficacité de ZYVOX 600 mg et d'autres médicaments antibactériens, ZYVOX doit être utilisé uniquement pour traiter ou prévenir les infections dont il est prouvé ou fortement suspecté qu'elles sont causées par des bactéries sensibles. Lorsque des informations sur la culture et la sensibilité sont disponibles, elles doivent être prises en compte lors de la sélection ou de la modification du traitement antibactérien. En l'absence de telles données, l'épidémiologie locale et les schémas de sensibilité peuvent contribuer à la sélection empirique du traitement.
DOSAGE ET ADMINISTRATION
Posologie générale et administration
La posologie recommandée pour les formulations de ZYVOX 600 mg pour le traitement des infections est décrite dans le tableau 1.
Aucun ajustement posologique n'est nécessaire lors du passage de l'administration intraveineuse à l'administration orale.
Administration intraveineuse
ZYVOX 600mg IV Injection est fourni dans des poches de perfusion à dose unique prêtes à l'emploi. Les produits médicamenteux parentéraux doivent être inspectés visuellement pour détecter les particules avant l'administration. Vérifiez s'il y a de petites fuites en pressant fermement le sac. Si des fuites sont détectées, jetez la solution, car la stérilité peut être altérée. Conservez les poches de perfusion dans le suremballage jusqu'à leur utilisation. Ranger à température ambiante. Protéger du gel. ZYVOX 600mg IV Injection peut présenter une couleur jaune qui peut s'intensifier avec le temps sans nuire à la puissance.
ZYVOX IV Injection doit être administré par perfusion intraveineuse sur une période de 30 à 120 minutes. Ne pas utiliser cette poche de perfusion intraveineuse dans des connexions en série. Aucun additif ne doit être introduit dans cette solution. Si ZYVOX 600 mg injection IV doit être administré en même temps qu'un autre médicament, chaque médicament doit être administré séparément conformément à la posologie et à la voie d'administration recommandées pour chaque produit. Jeter la portion inutilisée.
Si la même ligne intraveineuse est utilisée pour la perfusion séquentielle de plusieurs médicaments, la ligne doit être rincée avant et après la perfusion de ZYVOX IV Injection avec une solution de perfusion compatible avec ZYVOX 600 mg IV Injection et avec tout autre médicament administré via cette ligne commune. .
Compatibilités
Les solutions intraveineuses compatibles comprennent le chlorure de sodium injectable à 0,9 %, USP, le dextrose injectable à 5 %, USP et le lactate de Ringer injectable, USP.
Incompatibilités
Des incompatibilités physiques ont résulté de l'association de ZYVOX 600 mg IV injectable avec les médicaments suivants lors d'une administration simulée en Y : amphotéricine B, chlorpromazine HCl, diazépam, pentamidine isothionate, érythromycine lactobionate, phénytoïne sodique et triméthoprime-sulfaméthoxazole. De plus, une incompatibilité chimique s'est produite lorsque ZYVOX IV Injection a été associé à la ceftriaxone sodique.
Constitution de la suspension orale
ZYVOX pour suspension buvable est fourni sous forme de poudre/granule pour la constitution. Tapotez doucement le flacon pour détacher la poudre. Ajouter un total de 123 ml d'eau distillée en deux portions. Après avoir ajouté la première moitié, secouez vigoureusement pour mouiller toute la poudre. Ajouter ensuite la seconde moitié de l'eau et agiter vigoureusement pour obtenir une suspension homogène. Après constitution, chaque 5 mL de la suspension contient 100 mg de linézolide. Avant utilisation, mélanger délicatement en retournant le flacon 3 à 5 fois. Ne secouez pas. Conserver la suspension constituée à température ambiante. Utiliser dans les 21 jours après constitution.
COMMENT FOURNIE
Formes posologiques et points forts
ZYVOX 600mg IV Injection : 200 mg/100 mL (2 mg/mL) et 600 mg/300 mL (2 mg/mL) de linézolide à dose unique, sachets de perfusion en plastique souple prêts à l'emploi dans un suremballage en aluminium laminé.
Comprimé ZYVOX 600 mg :
Comprimé pelliculé blanc, en forme de capsule, gravé « ZYV » sur une face et « 600 » sur l'autre ZYVOX 600 mg pour suspension orale : granulé/poudre sec, blanc à blanc cassé, à saveur d'orange. Lorsqu'il est constitué selon les instructions, chaque flacon contiendra 150 ml d'une suspension fournissant l'équivalent de 100 mg de linézolide par 5 ml.
Stockage et manutention
Injection
ZYVOX 600mg IV L'injection est disponible dans des poches de perfusion en plastique flexibles à dose unique, prêtes à l'emploi, dans un suremballage en aluminium laminé. Les sacs et ports de perfusion ne sont pas fabriqués avec du latex de caoutchouc naturel. Les poches de perfusion sont disponibles dans les tailles d'emballage suivantes :
200 mg/100 mL (2 mg/mL) linézolide x 10 - CDN 0009-5137-04 600 mg/300 mL (2 mg/mL) linézolide x 10 - CDN 0009-5140-04
Comprimés
ZYVOX 600mg Comprimés sont disponibles comme suit :
600 mg (comprimés pelliculés blancs, en forme de capsule, gravés « ZYV » d'un côté et « 600 » de l'autre)
20 comprimés en flacon HDPE - CDN 0009-5138-02 Conditionnements unitaires de 30 comprimés - CDN 0009-5138-03
Suspension orale
ZYVOX pour suspension orale est disponible sous forme de granule/poudre sec, blanc à blanc cassé, aromatisé à l'orange. Lorsqu'il est constitué selon les instructions, chaque flacon contiendra 150 ml d'une suspension fournissant l'équivalent de 100 mg de linézolide par 5 ml. ZYVOX pour suspension orale est fourni comme suit :
100 mg/5 mL en flacons verre de 240 mL - CDN 0009-5136-01
Stockage et manutention
Conserver à 25 °C (77 °F). Protéger de la lumière. Conserver les flacons bien fermés pour les protéger de l'humidité. Il est recommandé de conserver les poches de perfusion dans le suremballage jusqu'à leur utilisation. Protéger les poches de perfusion du gel.
Distribué par Pharmacia & Upjohn Co Division de Pfizer Inc, NY, NY 10017. Révisé : novembre 2021.
EFFETS SECONDAIRES
Expérience des essais cliniques
Étant donné que les essais cliniques sont menés dans des conditions très variables, les taux d'effets indésirables observés dans les essais cliniques d'un médicament ne peuvent pas être directement comparés aux taux des essais cliniques d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique.
Adultes
L'innocuité des formulations de ZYVOX à 600 mg a été évaluée chez 2 046 patients adultes inscrits dans sept essais cliniques de phase 3 contrôlés par comparateur, qui ont été traités jusqu'à 28 jours.
Parmi les patients traités pour des infections non compliquées de la peau et des annexes cutanées (uSSSI), 25,4 % des patients traités par ZYVOX et 19,6 % des patients traités par un comparateur ont présenté au moins un événement indésirable lié au médicament. Pour toutes les autres indications, 20,4 % des patients traités par ZYVOX et 14,3 % des patients traités par le comparateur ont présenté au moins un événement indésirable lié au médicament.
Le tableau 2 montre l'incidence des effets indésirables toutes causes confondues liés au traitement signalés chez au moins 1 % des patients adultes dans ces essais par dose de ZYVOX.
Parmi les patients traités pour des uSSSI, 3,5 % des patients traités par ZYVOX et 2,4 % des patients traités par un comparateur ont arrêté le traitement en raison d'événements indésirables liés au médicament. Pour toutes les autres indications, des arrêts en raison d'événements indésirables liés au médicament sont survenus chez 2,1 % des patients traités par ZYVOX et 1,7 % des patients traités par le comparateur. Les événements indésirables liés au médicament les plus fréquemment signalés et ayant entraîné l'arrêt du traitement étaient les nausées, les maux de tête, la diarrhée et les vomissements.
Patients pédiatriques
La sécurité des formulations de ZYVOX a été évaluée chez 215 patients pédiatriques âgés de la naissance à 11 ans et chez 248 patients pédiatriques âgés de 5 à 17 ans (146 de ces 248 étaient âgés de 5 à 11 ans et 102 étaient âgés de 12 à 17 ans). Ces patients ont été inclus dans deux essais cliniques de phase 3 contrôlés par des comparateurs et ont été traités jusqu'à 28 jours. Dans l'étude portant sur des patients pédiatriques hospitalisés (de la naissance à 11 ans) atteints d'infections à Gram positif, qui ont été randomisés 2 contre 1 (linézolide : vancomycine), la mortalité était de 6,0 % (13/215) dans le bras linézolide et de 3,0 % (3/101) dans le bras vancomycine. Cependant, étant donné la maladie sous-jacente grave dans la population de patients, aucune causalité n'a pu être établie.
Parmi les patients pédiatriques traités pour des uSSSI, 19,2 % des patients traités par ZYVOX et 14,1 % des patients traités par un comparateur ont présenté au moins un événement indésirable lié au médicament. Pour toutes les autres indications, 18,8 % des patients traités par ZYVOX et 34,3 % des patients traités par le comparateur ont présenté au moins un événement indésirable lié au médicament.
Le tableau 3 montre l'incidence des effets indésirables toutes causes confondues liés au traitement signalés chez plus de 1 % des patients pédiatriques (et plus d'un patient) dans l'un ou l'autre groupe de traitement dans les essais de phase 3 contrôlés par comparateur.
Parmi les patients pédiatriques traités pour des uSSSI, 1,6 % des patients traités par ZYVOX et 2,4 % des patients traités par un comparateur ont arrêté le traitement en raison d'événements indésirables liés au médicament. Pour toutes les autres indications, des interruptions dues à des événements indésirables liés au médicament sont survenues chez 0,9 % des patients traités par ZYVOX et 6,1 % des patients traités par le comparateur.
Anomalies de laboratoire
ZYVOX a été associé à une thrombocytopénie lorsqu'il est utilisé à des doses allant jusqu'à 600 mg toutes les 12 heures pendant 28 jours maximum. Dans les essais contrôlés par comparateur de phase 3, le pourcentage de patients adultes ayant développé une numération plaquettaire substantiellement faible (définie comme moins de 75 % de la limite inférieure de la normale et/ou de la ligne de base) était de 2,4 % (intervalle entre les études : 0,3 à 10,0 %) avec ZYVOX 600 mg et 1,5 % (intervalle entre les études : 0,4 à 7,0 %) avec un comparateur. Dans une étude portant sur des patients pédiatriques hospitalisés dont l'âge s'étend de la naissance à 11 ans, le pourcentage de patients ayant développé une numération plaquettaire sensiblement faible (définie comme moins de 75 % de la limite inférieure de la normale et/ou de la ligne de base) était de 12,9 % avec ZYVOX 600 mg et 13,4 % avec la vancomycine. Dans une étude ambulatoire portant sur des patients pédiatriques âgés de 5 à 17 ans, le pourcentage de patients ayant développé une numération plaquettaire substantiellement faible était de 0 % avec ZYVOX et de 0,4 % avec le céfadroxil. La thrombocytopénie associée à l'utilisation de ZYVOX semble dépendre de la durée du traitement (généralement supérieure à 2 semaines de traitement). La numération plaquettaire de la plupart des patients est revenue à la normale/à la ligne de base au cours de la période de suivi. Aucun effet indésirable clinique lié n'a été identifié dans les essais cliniques de phase 3 chez des patients développant une thrombocytopénie. Des événements hémorragiques ont été identifiés chez des patients thrombocytopéniques dans le cadre d'un programme d'utilisation compassionnelle de ZYVOX ; le rôle du linézolide dans ces événements ne peut être déterminé [voir AVERTISSEMENTS ET PRECAUTIONS ].
Les changements observés dans d'autres paramètres de laboratoire, sans égard à la relation médicamenteuse, n'ont révélé aucune différence substantielle entre ZYVOX 600 mg et les comparateurs. Ces modifications n'étaient généralement pas cliniquement significatives, n'entraînaient pas l'arrêt du traitement et étaient réversibles. L'incidence des patients adultes et pédiatriques présentant au moins une valeur hématologique ou chimique sérique substantiellement anormale est présentée dans les tableaux 4, 5, 6 et 7.
Expérience post-commercialisation
Les effets indésirables suivants ont été identifiés lors de l'utilisation post-approbation de ZYVOX. Étant donné que ces réactions sont signalées volontairement à partir d'une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale avec l'exposition au médicament :
- Myélosuppression (y compris anémie, leucopénie, pancytopénie et thrombocytopénie) [voir AVERTISSEMENTS ET PRECAUTIONS ] ; anémie sidéroblastique.
- Neuropathie périphérique et neuropathie optique évoluant parfois vers une perte de vision [voir AVERTISSEMENTS ET PRECAUTIONS ].
- Acidose lactique [voir AVERTISSEMENTS ET PRECAUTIONS ]. Bien que ces rapports concernent principalement des patients traités pendant une durée supérieure à la durée maximale recommandée de 28 jours, ces événements ont également été rapportés chez des patients recevant des traitements plus courts.
- Un syndrome sérotoninergique a été rapporté chez des patients recevant des agents sérotoninergiques concomitants, y compris des antidépresseurs tels que les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS) et des opioïdes, et ZYVOX [voir AVERTISSEMENTS ET PRECAUTIONS ].
- Convulsions [voir AVERTISSEMENTS ET PRECAUTIONS ].
- Anaphylaxie, œdème de Quincke, affections cutanées bulleuses, y compris les effets indésirables cutanés graves (SCAR) tels que la nécrolyse épidermique toxique et le syndrome de Stevens-Johnson, et la vascularite d'hypersensibilité.
- Une décoloration superficielle des dents et une décoloration de la langue ont été rapportées avec l'utilisation du linézolide. La décoloration des dents a été éliminée avec un nettoyage dentaire professionnel (détartrage manuel) dans les cas dont l'issue était connue.
- Hypoglycémie, y compris épisodes symptomatiques [voir AVERTISSEMENTS ET PRECAUTIONS ].
- Hyponatrémie et/ou syndrome de sécrétion inappropriée d'hormone antidiurétique (SIADH) [voir AVERTISSEMENTS ET PRECAUTIONS ].
INTERACTIONS MÉDICAMENTEUSES
Inhibiteurs de la monoamine oxydase
Le linézolide est un inhibiteur réversible et non sélectif de la monoamine oxydase [voir CONTRE-INDICATIONS et PHARMACOLOGIE CLINIQUE ].
Agents adrénergiques et sérotoninergiques
Le linézolide peut interagir avec les agents adrénergiques et sérotoninergiques [voir AVERTISSEMENTS ET PRECAUTIONS et PHARMACOLOGIE CLINIQUE ].
AVERTISSEMENTS
Inclus dans le cadre du "PRÉCAUTIONS" Section
PRÉCAUTIONS
Myélosuppression
Une myélosuppression (incluant anémie, leucopénie, pancytopénie et thrombocytopénie) a été rapportée chez des patients recevant du linézolide. Dans les cas où le résultat est connu, lorsque le linézolide a été arrêté, les paramètres hématologiques affectés ont augmenté vers les niveaux de prétraitement. Une numération globulaire complète doit être surveillée chaque semaine chez les patients qui reçoivent du linézolide, en particulier chez ceux qui reçoivent du linézolide pendant plus de deux semaines, ceux qui présentent une myélosuppression préexistante, ceux qui reçoivent des médicaments concomitants qui produisent une suppression de la moelle osseuse ou ceux qui ont une infection chronique et qui ont reçu un traitement antibactérien antérieur ou concomitant. L'arrêt du traitement par le linézolide doit être envisagé chez les patients qui développent ou présentent une aggravation de la myélosuppression.
Neuropathie périphérique et optique
Des neuropathies périphériques et optiques ont été rapportées chez des patients traités par ZYVOX 600 mg, principalement chez les patients traités pendant une durée supérieure à la durée maximale recommandée de 28 jours. Dans les cas de neuropathie optique évoluant vers une perte de vision, les patients ont été traités pendant des périodes prolongées au-delà de la durée maximale recommandée. Des troubles visuels ont été signalés chez certains patients traités par ZYVOX pendant moins de 28 jours. Des neuropathies périphériques et optiques ont également été rapportées chez les enfants.
Si les patients présentent des symptômes de déficience visuelle, tels que des modifications de l'acuité visuelle, des modifications de la vision des couleurs, une vision floue ou une anomalie du champ visuel, une évaluation ophtalmique rapide est recommandée. La fonction visuelle doit être surveillée chez tous les patients prenant ZYVOX pendant des périodes prolongées (≥ 3 mois) et chez tous les patients signalant de nouveaux symptômes visuels, quelle que soit la durée du traitement par ZYVOX. En cas de survenue d'une neuropathie périphérique ou optique, la poursuite de l'utilisation de ZYVOX 600 mg chez ces patients doit être mise en balance avec les risques potentiels.
Syndrome sérotoninergique
Des rapports spontanés de syndrome sérotoninergique, y compris des cas mortels associés à la co-administration de ZYVOX et d'agents sérotoninergiques, y compris des antidépresseurs tels que les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS), ont été rapportés.
À moins que cela ne soit cliniquement approprié et que les patients ne soient étroitement surveillés pour détecter des signes et/ou des symptômes de syndrome sérotoninergique ou de réactions de type syndrome malin des neuroleptiques (de type SMN), le linézolide ne doit pas être administré aux patients atteints du syndrome carcinoïde et/ou aux patients prenant l'un des médicaments suivants : inhibiteurs de la recapture de la sérotonine, antidépresseurs tricycliques, bupropion, buspirone, agonistes des récepteurs 5-HT1 de la sérotonine (triptans) et opioïdes, y compris la mépéridine [voir INTERACTIONS MÉDICAMENTEUSES et PHARMACOLOGIE CLINIQUE ].
Dans certains cas, un patient recevant déjà un antidépresseur sérotoninergique ou de la buspirone peut nécessiter un traitement urgent par le linézolide. Si des alternatives au linézolide ne sont pas disponibles et que les bénéfices potentiels du linézolide l'emportent sur les risques de syndrome sérotoninergique ou de réactions de type SMN, l'antidépresseur sérotoninergique doit être arrêté rapidement et le linézolide doit être administré. Le patient doit être surveillé pendant deux semaines (cinq semaines si la fluoxétine a été prise) ou jusqu'à 24 heures après la dernière dose de linézolide, selon la première éventualité. Les symptômes du syndrome sérotoninergique ou des réactions de type SMN comprennent l'hyperthermie, la rigidité, les myoclonies, l'instabilité autonome et les modifications de l'état mental qui incluent une agitation extrême évoluant vers le délire et le coma. Le patient doit également être surveillé pour détecter les symptômes de sevrage de l'antidépresseur (voir la notice du ou des agents spécifiés pour une description des symptômes de sevrage associés).
Déséquilibre de la mortalité dans une étude expérimentale chez des patients atteints de bactériémies liées au cathéter, y compris ceux atteints d'infections au site du cathéter
Un déséquilibre de la mortalité a été observé chez les patients traités par le linézolide par rapport à la vancomycine/dicloxacilline/oxacilline dans une étude en ouvert chez des patients gravement malades avec des infections liées aux cathéters intravasculaires [78/363 (21,5 %) contre 58/363 (16,0 % ); rapport de cotes 1,426, IC à 95 % 0,970, 2,098]. Bien que la causalité n'ait pas été établie, ce déséquilibre observé s'est produit principalement chez les patients traités par linézolide chez lesquels soit des pathogènes Gram-négatifs, des pathogènes mixtes Gram-négatifs et Gram-positifs, soit aucun pathogène n'ont été identifiés au départ, mais n'a pas été observé chez les patients avec Infections à Gram positif uniquement.
Le linézolide n'est pas approuvé et ne doit pas être utilisé pour le traitement des patients atteints d'infections de la circulation sanguine liées au cathéter ou d'infections au site du cathéter.
Le linézolide n'a aucune activité clinique contre les pathogènes à Gram négatif et n'est pas indiqué pour le traitement des infections à Gram négatif. Il est essentiel qu'un traitement Gram négatif spécifique soit initié immédiatement si un agent pathogène Gram négatif concomitant est documenté ou suspecté [voir LES INDICATIONS ].
Diarrhée associée à Clostridioides Difficile
Des diarrhées associées à Clostridioides difficile (CDAD) ont été signalées avec l'utilisation de presque tous les agents antibactériens, y compris ZYVOX 600 mg, et leur gravité peut varier d'une diarrhée légère à une colite mortelle. Le traitement avec des agents antibactériens altère la flore normale du côlon, entraînant une prolifération de C. difficile.
C. difficile produit les toxines A et B qui contribuent au développement de CDAD. Les souches de C. difficile productrices d'hypertoxines entraînent une morbidité et une mortalité accrues, car ces infections peuvent être réfractaires au traitement antimicrobien et nécessiter une colectomie. La DACD doit être envisagée chez tous les patients qui présentent une diarrhée suite à l'utilisation de médicaments antibactériens.
Des antécédents médicaux minutieux sont nécessaires car il a été rapporté que la DACD survient plus de deux mois après l'administration d'agents antibactériens.
Si la DACD est suspectée ou confirmée, l'utilisation continue de médicaments antibactériens non dirigés contre C. difficile peut devoir être interrompue. Une gestion appropriée des fluides et des électrolytes, une supplémentation en protéines, un traitement médicamenteux antibactérien de C. difficile et une évaluation chirurgicale doivent être institués selon les indications cliniques.
Interactions potentielles produisant une élévation de la tension artérielle
À moins que les patients ne soient surveillés pour des augmentations potentielles de la pression artérielle, le linézolide ne doit pas être administré aux patients souffrant d'hypertension non contrôlée, de phéochromocytome, de thyrotoxicose et/ou aux patients prenant l'un des types de médicaments suivants : agents sympathomimétiques à action directe et indirecte (p. ex. pseudoéphédrine), agents vasopresseurs (p. ex., épinéphrine, noradrénaline), agents dopaminergiques (p. ex., dopamine, dobutamine) [voir INTERACTIONS MÉDICAMENTEUSES et PHARMACOLOGIE CLINIQUE ].
Acidose lactique
Une acidose lactique a été rapportée avec l'utilisation de ZYVOX. Dans les cas rapportés, les patients ont présenté des épisodes répétés de nausées et de vomissements. Les patients qui développent des nausées ou des vomissements récurrents, une acidose inexpliquée ou un faible taux de bicarbonate pendant qu'ils reçoivent ZYVOX doivent recevoir une évaluation médicale immédiate.
Convulsions
Des convulsions ont été rapportées chez des patients traités par le linézolide. Dans certains de ces cas, des antécédents de convulsions ou des facteurs de risque de convulsions ont été signalés.
Hypoglycémie
Des cas d'hypoglycémie symptomatique post-commercialisation ont été rapportés chez des patients atteints de diabète sucré recevant de l'insuline ou des hypoglycémiants oraux lorsqu'ils étaient traités par le linézolide, un inhibiteur de la MAO réversible et non sélectif. Certains inhibiteurs de la MAO ont été associés à des épisodes hypoglycémiques chez des patients diabétiques recevant de l'insuline ou des agents hypoglycémiants. Bien qu'une relation causale entre le linézolide et l'hypoglycémie n'ait pas été établie, les patients diabétiques doivent être avertis des réactions hypoglycémiques potentielles lorsqu'ils sont traités par le linézolide.
En cas d'hypoglycémie, une diminution de la dose d'insuline ou d'hypoglycémiant oral, ou l'arrêt de l'hypoglycémiant oral, de l'insuline ou du linézolide peut être nécessaire.
Hyponatrémie et/ou syndrome de sécrétion inappropriée d'hormone antidiurétique (SIADH)
Des cas post-commercialisation d'hyponatrémie et/ou de syndrome de sécrétion inappropriée d'hormone antidiurétique (SIADH) ont été observés chez des patients traités par le linézolide. Dans les cas signalés, les signes et symptômes incluaient la confusion, la somnolence, une faiblesse généralisée et, dans les cas graves, une insuffisance respiratoire et même la mort. Surveillez régulièrement les taux sériques de sodium chez les personnes âgées, chez les patients prenant des diurétiques et chez les autres patients à risque d'hyponatrémie et/ou de SIADH pendant la prise de ZYVOX. Si des signes et des symptômes d'hyponatrémie et/ou de SIADH apparaissent, arrêtez ZYVOX 600 mg et mettez en place des mesures de soutien appropriées.
Risques chez les patients atteints de phénylcétonurie
La phénylalanine peut être nocive pour les patients atteints de phénylcétonurie (PCU). ZYVOX pour suspension buvable contient de la phénylalanine, un composant de l'aspartame. Chaque 5 mL de suspension buvable à 100 mg/5 mL contient 20 mg de phénylalanine. Avant de prescrire ZYVOX 600 mg pour suspension buvable à un patient atteint de PCU, tenez compte de la quantité quotidienne combinée de phénylalanine de toutes les sources, y compris ZYVOX pour suspension buvable.
Les autres formulations de ZYVOX ne contiennent pas de phénylalanine.
Développement de bactéries résistantes aux médicaments
La prescription de ZYVOX en l'absence d'infection bactérienne avérée ou fortement suspectée ou d'indication prophylactique est peu susceptible d'apporter un bénéfice au patient et augmente le risque de développement de bactéries résistantes aux médicaments.
Toxicologie non clinique
Carcinogenèse, mutagenèse, altération de la fertilité
Aucune étude sur la durée de vie chez l'animal n'a été menée pour évaluer le potentiel carcinogène du linézolide. Aucun potentiel mutagène ou clastogène n'a été trouvé dans une batterie de tests comprenant : des tests de mutagénicité (réversion bactérienne d'Ames et mutation des cellules CHO), un test de synthèse d'ADN non programmée (UDS) in vitro, un test d'aberration chromosomique in vitro dans des lymphocytes humains et un test du micronoyau de souris in vivo.
Le linézolide n'a pas affecté la fertilité ou les performances de reproduction des rats femelles adultes ayant reçu des doses orales allant jusqu'à 100 mg/kg/jour pendant 14 jours avant l'accouplement jusqu'au jour 7 de la gestation. Il a diminué de manière réversible la fertilité et les performances de reproduction chez les rats mâles adultes lorsqu'il a été administré à doses ≥ 50 mg/kg/jour, avec des expositions approximativement égales ou supérieures au niveau d'exposition humaine attendu (les comparaisons d'exposition sont basées sur les ASC). Les effets réversibles sur la fertilité ont été médiés par une spermatogenèse altérée. Les spermatides affectées contenaient des mitochondries anormalement formées et orientées et n'étaient pas viables. Une hypertrophie des cellules épithéliales et une hyperplasie de l'épididyme ont été observées en conjonction avec une diminution de la fertilité. Des changements épididymaires similaires n'ont pas été observés chez les chiens.
Chez les rats mâles sexuellement matures exposés au médicament à l'âge juvénile, une légère diminution de la fertilité a été observée après traitement par le linézolide pendant la majeure partie de leur période de développement sexuel (50 mg/kg/jour du 7e au 36e jour et 100 mg/kg/jour de l'âge de 37 à 55 jours), avec des expositions jusqu'à 1,7 fois supérieures aux ASC moyennes observées chez les patients pédiatriques âgés de 3 mois à 11 ans. Aucune diminution de la fertilité n'a été observée avec des périodes de traitement plus courtes, correspondant à une exposition in utero pendant la période néonatale précoce (jour de gestation 6 à jour postnatal 5), à une exposition néonatale (jours postnatals 5 à 21) ou à une exposition juvénile (jours postnatals 22 à 35 ). Des réductions réversibles de la motilité des spermatozoïdes et une altération de la morphologie des spermatozoïdes ont été observées chez les rats traités du 22e au 35e jour postnatal.
Utilisation dans des populations spécifiques
Grossesse
Résumé des risques
Les données disponibles provenant de rapports de cas publiés et post-commercialisation sur l'utilisation du linézolide chez les femmes enceintes n'ont pas identifié de risque associé au médicament d'anomalies congénitales majeures, de fausse couche ou d'issues maternelles ou fœtales indésirables. Lorsqu'il a été administré pendant l'organogenèse, le linézolide n'a pas provoqué de malformations chez la souris, le rat ou le lapin à des niveaux d'exposition maternelle d'environ 6,5 fois (souris), équivalents à (rats) ou 0,06 fois (lapins) l'exposition thérapeutique clinique, sur la base des ASC. Cependant, une létalité embryo-fœtale a été observée chez la souris à 6,5 fois l'exposition humaine estimée. Lorsque des rats femelles ont reçu une dose pendant l'organogenèse jusqu'à la lactation, la survie postnatale des ratons a diminué à des doses approximativement équivalentes à l'exposition humaine estimée sur la base des ASC (voir Données ).
Le risque de fond de malformations congénitales majeures et de fausse couche pour les populations indiquées est inconnu. Toutes les grossesses ont un risque de fond de malformation congénitale, de perte ou d'autres résultats indésirables. Dans la population générale des États-Unis, le risque de fond estimé de malformations congénitales majeures et de fausse couche dans les grossesses cliniquement reconnues est de 2 à 4 % et de 15 à 20 %, respectivement.
Données
Données animales
Chez la souris, des toxicités embryo-fœtales n'ont été observées qu'à des doses entraînant une toxicité maternelle (signes cliniques et diminution de la prise de poids corporel). Une dose orale de 450 mg/kg/jour administrée du 6e au 16e jour de gestation (JG) (6,5 fois l'exposition humaine estimée sur la base des ASC) était corrélée à une augmentation de la mortalité embryonnaire post-implantation, y compris la perte totale de la portée, une diminution du poids corporel des fœtus et une augmentation de l'incidence de la fusion du cartilage costal. Aucune toxicité maternelle ou embryo-fœtale n'a été observée à des doses allant jusqu'à 150 mg/kg/jour. Aucune malformation fœtale n'a été observée.
Chez le rat, une toxicité fœtale a été observée à 15 et 50 mg/kg/jour administrés par voie orale du 6e au 17e jour (expositions 0,22 fois à environ équivalentes à l'exposition humaine estimée, respectivement, sur la base des ASC). Les effets consistaient en une diminution du poids corporel du fœtus et une réduction de l'ossification des sternèbres, un résultat souvent observé en association avec une diminution du poids corporel du fœtus. Aucune malformation fœtale n'a été observée. Une toxicité maternelle, sous la forme d'une réduction du gain de poids corporel, a été observée à 50 mg/kg/jour.
Chez les lapins, une réduction du poids corporel fœtal n'est survenue qu'en présence d'une toxicité maternelle (signes cliniques, réduction du gain de poids corporel et de la consommation alimentaire) lorsqu'il a été administré à une dose orale de 15 mg/kg/jour administrée du 6e au 20e jour (0,06 fois la exposition humaine estimée sur la base des ASC). Aucune malformation fœtale n'a été observée.
Lorsque des rats femelles ont été traitées avec 50 mg/kg/jour (environ l'équivalent de l'exposition humaine estimée sur la base des ASC) de linézolide pendant la grossesse et l'allaitement (JG 6 au jour de lactation 20), la survie des ratons a diminué les jours postnatals 1 à 4 Les petits mâles et femelles autorisés à atteindre l'âge de reproduction, lorsqu'ils sont accouplés, ont montré une augmentation de la perte préimplantatoire.
Lactation
Résumé des risques
Le linézolide est présent dans le lait maternel. D'après les données des rapports de cas publiés disponibles, la dose quotidienne de linézolide que le nourrisson recevrait du lait maternel serait d'environ 6 % à 9 % de la dose thérapeutique recommandée pour le nourrisson (10 mg/kg toutes les 8 heures). Il n'existe aucune information sur les effets du linézolide sur le nourrisson allaité ; cependant, la diarrhée et les vomissements étaient les effets indésirables les plus fréquemment rapportés dans les essais cliniques chez les nourrissons recevant du linézolide à titre thérapeutique [voir EFFETS INDÉSIRABLES et (voir Considérations cliniques). Il n'existe aucune information sur les effets du linézolide sur la production de lait. Les avantages de l'allaitement pour le développement et la santé doivent être pris en compte, ainsi que le besoin clinique de linézolide de la mère et tout effet indésirable potentiel sur l'enfant allaité du linézolide ou de l'affection maternelle sous-jacente.
Considérations cliniques
Conseillez aux femmes qui allaitent de surveiller un nourrisson allaité pour la diarrhée et les vomissements.
Femelles et mâles de potentiel reproducteur
Infertilité
Mâles
D'après les résultats d'études chez le rat, ZYVOX peut altérer de manière réversible la fertilité chez les patients de sexe masculin [voir Toxicologie non clinique )].
Utilisation pédiatrique
L'innocuité et l'efficacité de ZYVOX 600 mg pour le traitement des patients pédiatriques atteints des infections suivantes sont étayées par des preuves issues d'études adéquates et bien contrôlées chez l'adulte, des données pharmacocinétiques chez des patients pédiatriques et des données supplémentaires provenant d'une étude contrôlée par comparateur de Gram-positif infections chez les patients pédiatriques âgés de la naissance à 11 ans [voir LES INDICATIONS , PHARMACOLOGIE CLINIQUE et Etudes cliniques ] :
- pneumonie nosocomiale
- infections compliquées de la peau et des tissus cutanés
- pneumonie communautaire (également étayée par des preuves d'une étude non contrôlée chez des patients âgés de 8 mois à 12 ans)
- infections à Enterococcus faecium résistant à la vancomycine
La sécurité et l'efficacité de ZYVOX 600 mg pour le traitement des patients pédiatriques atteints de l'infection suivante ont été établies dans une étude contrôlée par comparateur chez des patients pédiatriques âgés de 5 à 17 ans [voir Etudes cliniques ] :
- infections non compliquées de la peau et des structures cutanées causées par Staphylococcus aureus (souches sensibles à la méthicilline uniquement) ou Streptococcus pyogenes
Les informations pharmacocinétiques générées chez les patients pédiatriques présentant des shunts ventriculopéritonéaux ont montré des concentrations variables de linézolide dans le liquide céphalo-rachidien (LCR) après une administration unique et multiple de linézolide ; les concentrations thérapeutiques n'étaient pas systématiquement atteintes ou maintenues dans le LCR. Par conséquent, l'utilisation du linézolide pour le traitement empirique des patients pédiatriques atteints d'infections du système nerveux central n'est pas recommandée.
La pharmacocinétique du linézolide a été évaluée chez des patients pédiatriques de la naissance à 17 ans. En général, la clairance du linézolide en fonction du poids diminue progressivement avec l'âge des patients pédiatriques. Cependant, chez les nouveau-nés prématurés (âge gestationnel DOSAGE ET ADMINISTRATION et PHARMACOLOGIE CLINIQUE ].
Dans une expérience clinique limitée, 5 patients pédiatriques sur 6 (83 %) présentant des infections dues à des agents pathogènes à Gram positif avec des concentrations minimales inhibitrices (CMI) de 4 mcg/mL traités avec ZYVOX 600 mg ont eu des guérisons cliniques. Cependant, les patients pédiatriques présentent une plus grande variabilité de la clairance du linézolide et de l'exposition systémique (ASC) par rapport aux adultes. Chez les patients pédiatriques présentant une réponse clinique sous-optimale, en particulier ceux présentant des agents pathogènes avec une CMI de 4 mcg/mL, une exposition systémique, un site et une gravité de l'infection plus faibles, ainsi que la condition médicale sous-jacente, doivent être pris en compte lors de l'évaluation de la réponse clinique [voir PHARMACOLOGIE CLINIQUE et DOSAGE ET ADMINISTRATION ].
Utilisation gériatrique
Sur les 2 046 patients traités par ZYVOX 600 mg dans les essais cliniques de phase 3 contrôlés par comparateur, 589 (29 %) avaient 65 ans ou plus et 253 (12 %) avaient 75 ans ou plus. Aucune différence globale d'innocuité ou d'efficacité n'a été observée entre ces patients et les patients plus jeunes, et d'autres expériences cliniques rapportées n'ont pas identifié de différences dans les réponses entre les patients âgés et les patients plus jeunes, mais une plus grande sensibilité de certains individus plus âgés ne peut être exclue.
SURDOSAGE
En cas de surdosage, des soins de support sont conseillés, avec maintien de la filtration glomérulaire. L'hémodialyse peut faciliter une élimination plus rapide du linézolide. Dans un essai clinique de phase 1, environ 30 % d'une dose de linézolide a été éliminée au cours d'une séance d'hémodialyse de 3 heures commençant 3 heures après l'administration de la dose de linézolide. Aucune donnée n'est disponible pour l'élimination du linézolide par dialyse péritonéale ou hémoperfusion. Les signes cliniques de toxicité aiguë chez les animaux étaient une diminution de l'activité et de l'ataxie chez les rats et des vomissements et des tremblements chez les chiens traités avec 3 000 mg/kg/jour et 2 000 mg/kg/jour, respectivement.
CONTRE-INDICATIONS
Hypersensibilité
Les formulations de ZYVOX sont contre-indiquées chez les patients présentant une hypersensibilité connue au linézolide ou à l'un des autres composants du produit.
Inhibiteurs de la monoamine oxydase
Le linézolide ne doit pas être utilisé chez les patients prenant un médicament qui inhibe les monoamine oxydases A ou B (par exemple, phénelzine, isocarboxazide) ou dans les deux semaines suivant la prise d'un tel médicament.
PHARMACOLOGIE CLINIQUE
Mécanisme d'action
ZYVOX est un médicament antibactérien [voir Microbiologie ].
Pharmacodynamie
Dans une étude approfondie de l'intervalle QT randomisée, positive et contrôlée par placebo, 40 sujets sains ont reçu une dose unique de 600 mg de ZYVOX via une perfusion IV d'une heure, une dose unique de 1 200 mg de ZYVOX via une perfusion IV d'une heure, un placebo et un dose orale unique de contrôle positif. Aux doses de 600 mg et de 1 200 mg de ZYVOX 600 mg, aucun effet significatif sur l'intervalle QTc n'a été détecté à la concentration plasmatique maximale ou à tout autre moment.
Pharmacocinétique
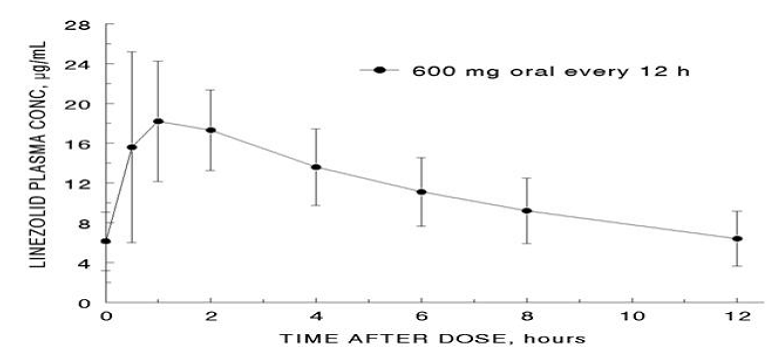
Les paramètres pharmacocinétiques moyens du linézolide chez l'adulte après des doses orales et intraveineuses uniques et multiples sont résumés dans le tableau 8. Les concentrations plasmatiques de linézolide à l'état d'équilibre après des doses orales de 600 mg administrées toutes les 12 heures sont présentées dans la figure 1.
Figure 1. Concentrations plasmatiques de linézolide chez les adultes à l'état d'équilibre après administration orale toutes les 12 heures (moyenne ± écart type, n = 16)
Absorption
Le linézolide est largement absorbé après administration orale. Les concentrations plasmatiques maximales sont atteintes environ 1 à 2 heures après l'administration et la biodisponibilité absolue est d'environ 100 %. Par conséquent, le linézolide peut être administré par voie orale ou intraveineuse sans ajustement posologique.
Le linézolide peut être administré sans tenir compte de l'heure des repas. Le temps nécessaire pour atteindre la concentration maximale est retardé de 1,5 heure à 2,2 heures et la Cmax est diminuée d'environ 17 % lorsque des aliments riches en graisses sont administrés avec du linézolide. Cependant, l'exposition totale mesurée comme AUC0-∞ est similaire dans les deux conditions.
Distribution
Des études pharmacocinétiques animales et humaines ont démontré que le linézolide se distribue facilement dans les tissus bien perfusés. La liaison aux protéines plasmatiques du linézolide est d'environ 31 % et est indépendante de la concentration. Le volume de distribution du linézolide à l'état d'équilibre était en moyenne de 40 à 50 litres chez des volontaires adultes sains.
Les concentrations de linézolide ont été déterminées dans divers liquides provenant d'un nombre limité de sujets lors d'études de phase 1 sur des volontaires après administration multiple de linézolide. Le rapport du linézolide dans la salive par rapport au plasma était de 1,2 à 1 et le rapport du linézolide dans la sueur par rapport au plasma était de 0,55 à 1.
Métabolisme
Le linézolide est principalement métabolisé par oxydation du cycle morpholine, ce qui donne deux métabolites acides carboxyliques à cycle ouvert inactifs : le métabolite acide aminoéthoxyacétique (A) et le métabolite hydroxyéthylglycine (B). La formation du métabolite A est supposée être formée via une voie enzymatique alors que le métabolite B est médié par un mécanisme d'oxydation chimique non enzymatique in vitro. Des études in vitro ont démontré que le linézolide est peu métabolisé et peut être médié par le cytochrome P450 humain. Cependant, la voie métabolique du linézolide n'est pas entièrement comprise.
Excrétion
La clairance non rénale représente environ 65 % de la clairance totale du linézolide. À l'état d'équilibre, environ 30 % de la dose apparaît dans l'urine sous forme de linézolide, 40 % sous forme de métabolite B et 10 % sous forme de métabolite A. La clairance rénale moyenne du linézolide est de 40 mL/min, ce qui suggère une réabsorption tubulaire nette. Pratiquement aucun linézolide n'apparaît dans les fèces, alors qu'environ 6 % de la dose apparaît dans les fèces sous forme de métabolite B et 3 % sous forme de métabolite A.
Un faible degré de non-linéarité de la clairance a été observé avec des doses croissantes de linézolide, ce qui semble être dû à une clairance rénale et non rénale plus faible du linézolide à des concentrations plus élevées. Cependant, la différence de clairance était faible et n'a pas été reflétée dans la demi-vie d'élimination apparente.
Populations spécifiques
Patients gériatriques
La pharmacocinétique du linézolide n'est pas significativement modifiée chez les patients âgés (65 ans ou plus). Par conséquent, l'adaptation de la dose pour les patients gériatriques n'est pas nécessaire.
Patients pédiatriques
La pharmacocinétique du linézolide après une dose intraveineuse unique a été étudiée chez des patients pédiatriques âgés de la naissance à 17 ans (y compris les nouveau-nés prématurés et nés à terme), chez des adolescents en bonne santé âgés de 12 à 17 ans et chez des patients pédiatriques âgés de dans l'âge de 1 semaine à 12 ans. Les paramètres pharmacocinétiques du linézolide sont résumés dans le tableau 9 pour les populations pédiatriques étudiées et les sujets adultes sains après administration de doses intraveineuses uniques.
La Cmax et le volume de distribution (Vss) du linézolide sont similaires quel que soit l'âge chez les patients pédiatriques. Cependant, la clairance plasmatique du linézolide varie en fonction de l'âge. À l'exclusion des nouveau-nés prématurés âgés de moins d'une semaine, la clairance basée sur le poids est la plus rapide dans les tranches d'âge les plus jeunes allant de
Des valeurs quotidiennes moyennes similaires de l'ASC ont été observées chez les patients pédiatriques de la naissance à 11 ans recevant toutes les 8 heures par rapport aux adolescents ou aux adultes recevant toutes les 12 heures. Par conséquent, la posologie pour les patients pédiatriques jusqu'à 11 ans doit être de 10 mg/kg toutes les 8 heures. Les patients pédiatriques de 12 ans et plus doivent recevoir 600 mg toutes les 12 heures [voir DOSAGE ET ADMINISTRATION ].
Le genre
Les femmes ont un volume de distribution du linézolide légèrement inférieur à celui des hommes. Les concentrations plasmatiques sont plus élevées chez les femmes que chez les hommes, ce qui est en partie dû aux différences de poids corporel. Après une dose de 600 mg, la clairance orale moyenne est environ 38 % plus faible chez les femmes que chez les hommes. Cependant, il n'y a pas de différences significatives entre les sexes dans la constante de taux d'élimination apparente moyenne ou la demi-vie. Ainsi, l'exposition aux médicaments chez les femmes ne devrait pas augmenter sensiblement au-delà des niveaux connus pour être bien tolérés. Par conséquent, un ajustement posologique selon le sexe ne semble pas nécessaire.
Insuffisance rénale
La pharmacocinétique de la molécule mère, le linézolide, n'est pas altérée chez les patients présentant un degré quelconque d'insuffisance rénale ; cependant, les deux principaux métabolites du linézolide s'accumulent chez les patients atteints d'insuffisance rénale, la quantité d'accumulation augmentant avec la sévérité de l'insuffisance rénale (voir Tableau 10). La pharmacocinétique du linézolide et de ses deux métabolites a également été étudiée chez des patients atteints d'insuffisance rénale terminale (IRT) sous hémodialyse. Dans l'étude ESRD, 14 patients ont reçu 600 mg de linézolide toutes les 12 heures pendant 14,5 jours (voir Tableau 11). Étant donné que des concentrations plasmatiques similaires de linézolide sont atteintes quelle que soit la fonction rénale, aucun ajustement posologique n'est recommandé chez les patients insuffisants rénaux. Cependant, compte tenu de l'absence d'informations sur la signification clinique de l'accumulation des métabolites primaires, l'utilisation du linézolide chez les insuffisants rénaux doit être mise en balance avec les risques potentiels d'accumulation de ces métabolites. Le linézolide et les deux métabolites sont éliminés par hémodialyse. Aucune information n'est disponible sur l'effet de la dialyse péritonéale sur la pharmacocinétique du linézolide. Environ 30 % d'une dose ont été éliminés au cours d'une séance d'hémodialyse de 3 heures commençant 3 heures après l'administration de la dose de linézolide; par conséquent, le linézolide doit être administré après l'hémodialyse.
Insuffisance hépatique
La pharmacocinétique du linézolide n'est pas altérée chez les patients (n=7) présentant une insuffisance hépatique légère à modérée (Child-Pugh classe A ou B). Sur la base des informations disponibles, aucun ajustement posologique n'est recommandé chez les patients présentant une insuffisance hépatique légère à modérée. La pharmacocinétique du linézolide chez les patients atteints d'insuffisance hépatique sévère n'a pas été évaluée.
Interactions médicamenteuses
Médicaments métabolisés par le cytochrome P450
Le linézolide n'est pas un inducteur du cytochrome P450 (CYP450) chez le rat. De plus, le linézolide n'inhibe pas les activités des isoformes humaines cliniquement significatives du CYP (p. ex., 1A2, 2C9, 2C19, 2D6, 2E1, 3A4). Par conséquent, le linézolide ne devrait pas affecter la pharmacocinétique des autres médicaments métabolisés par ces principales enzymes. L'administration concomitante de linézolide ne modifie pas sensiblement les caractéristiques pharmacocinétiques de la (S)-warfarine, qui est largement métabolisée par le CYP2C9. Des médicaments tels que la warfarine et la phénytoïne, qui sont des substrats du CYP2C9, peuvent être administrés avec le linézolide sans modification de la posologie.
Médicaments antibactériens
Aztréonam : La pharmacocinétique du linézolide ou de l'aztréonam n'est pas modifiée lorsqu'ils sont administrés ensemble.
Gentamicine : La pharmacocinétique du linézolide ou de la gentamicine n'est pas modifiée lorsqu'ils sont administrés ensemble.
Antioxydants
Le potentiel d'interactions médicamenteuses avec le linézolide et les antioxydants vitamine C et vitamine E a été étudié chez des volontaires sains. Les sujets ont reçu une dose orale de 600 mg de linézolide le jour 1 et une autre dose de 600 mg de linézolide le jour 8. Les jours 2 à 9, les sujets ont reçu soit de la vitamine C (1 000 mg/jour) soit de la vitamine E (800 UI/ journée). L'ASC0-∞ du linézolide a augmenté de 2,3 % lorsqu'il est co-administré avec de la vitamine C et de 10,9 % lorsqu'il est co-administré avec de la vitamine E. Aucun ajustement de la dose de linézolide n'est recommandé lors de la co-administration avec de la vitamine C ou de la vitamine E.
Inducteurs puissants du CYP 3A4
Rifampine
L'effet de la rifampicine sur la pharmacocinétique du linézolide a été évalué dans une étude portant sur 16 hommes adultes en bonne santé. Les volontaires ont reçu 600 mg de linézolide par voie orale deux fois par jour pendant 5 doses avec et sans rifampicine 600 mg une fois par jour pendant 8 jours. L'administration concomitante de rifampicine et de linézolide a entraîné une diminution de 21 % de la Cmax du linézolide [IC à 90 %, 15 % - 27 %] et une diminution de 32 % de l'ASC0-12 du linézolide [IC à 90 %, 27 % - 37 %]. La signification clinique de cette interaction est inconnue. Le mécanisme de cette interaction n'est pas entièrement compris et peut être lié à l'induction d'enzymes hépatiques. D'autres inducteurs puissants des enzymes hépatiques (par ex. carbamazépine, phénytoïne, phénobarbital) pourraient entraîner une diminution similaire ou moindre de l'exposition au linézolide.
Inhibition de la monoamine oxydase
Le linézolide est un inhibiteur réversible et non sélectif de la monoamine oxydase. Par conséquent, le linézolide peut interagir avec les agents adrénergiques et sérotoninergiques.
Agents adrénergiques
Certaines personnes recevant ZYVOX 600 mg peuvent présenter une amélioration réversible de la réponse pressive aux agents sympathomimétiques à action indirecte, aux vasopresseurs ou aux agents dopaminergiques. Des médicaments couramment utilisés tels que la phénylpropanolamine et la pseudoéphédrine ont été spécifiquement étudiés. Les doses initiales d'agents adrénergiques, tels que la dopamine ou l'épinéphrine, doivent être réduites et titrées pour obtenir la réponse souhaitée.
Tyramine
Une réponse pressive significative a été observée chez des sujets adultes normaux recevant des doses de linézolide et de tyramine supérieures à 100 mg. Par conséquent, les patients recevant du linézolide doivent éviter de consommer de grandes quantités d'aliments ou de boissons à forte teneur en tyramine [voir INFORMATIONS PATIENTS ].
Pseudoéphédrine HCl ou phénylpropanolamine HCl
Une amélioration réversible de la réponse pressive du chlorhydrate de pseudoéphédrine (PSE) ou du chlorhydrate de phénylpropanolamine (PPA) est observée lorsque le linézolide est administré à des sujets sains normotendus [voir AVERTISSEMENTS ET PRECAUTIONS et INTERACTIONS MÉDICAMENTEUSES ]. Une étude similaire n'a pas été menée chez des patients hypertendus. Les études d'interaction menées chez des sujets normotendus ont évalué les effets sur la pression artérielle et la fréquence cardiaque du placebo, du PPA ou du PSE seul, du linézolide seul et de l'association du linézolide à l'état d'équilibre (600 mg toutes les 12 heures pendant 3 jours) avec deux doses de PPA ( 25 mg) ou PSE (60 mg) à 4 heures d'intervalle. La fréquence cardiaque n'a été affectée par aucun des traitements. La pression artérielle a été augmentée avec les deux traitements combinés. Les niveaux de pression artérielle maximum ont été observés 2 à 3 heures après la deuxième dose de PPA ou de PSE, et sont revenus à la ligne de base 2 à 3 heures après le pic. Les résultats de l'étude PPA suivent, montrant la pression artérielle systolique maximale moyenne (et étendue) en mm Hg : placebo = 121 (103 à 158) ; linézolide seul = 120 (107 à 135) ; PPA seul = 125 (106 à 139) ; PPA avec linézolide = 147 (129 à 176). Les résultats de l'étude PSE étaient similaires à ceux de l'étude PPA. L'augmentation maximale moyenne de la pression artérielle systolique par rapport au départ était de 32 mm Hg (intervalle : 20-52 mm Hg) et de 38 mm Hg (intervalle : 18-79 mm Hg) lors de la co-administration de linézolide avec la pseudoéphédrine ou la phénylpropanolamine, respectivement.
Agents sérotoninergiques
Dextrométhorphane
L'interaction médicamenteuse potentielle avec le dextrométhorphane a été étudiée chez des volontaires sains. Les sujets ont reçu du dextrométhorphane (deux doses de 20 mg administrées à 4 heures d'intervalle) avec ou sans linézolide. Aucun effet du syndrome sérotoninergique (confusion, délire, agitation, tremblements, rougeurs, diaphorèse, hyperpyrexie) n'a été observé chez les sujets normaux recevant du linézolide et du dextrométhorphane.
Microbiologie
Mécanisme d'action
Le linézolide est un agent antibactérien synthétique de la classe des oxazolidinones, qui a une utilité clinique dans le traitement des infections causées par des bactéries aérobies à Gram positif. Le spectre d'activité in vitro du linézolide comprend également certaines bactéries Gram-négatives et des bactéries anaérobies. Le linézolide se lie à un site sur l'ARN ribosomal bactérien 23S de la sous-unité 50S et empêche la formation d'un complexe d'initiation 70S fonctionnel, essentiel à la reproduction bactérienne. Les résultats des études de destruction par le temps ont montré que le linézolide était bactériostatique contre les entérocoques et les staphylocoques. Pour les streptocoques, le linézolide s'est avéré bactéricide pour la majorité des isolats.
La résistance
Des études in vitro ont montré que des mutations ponctuelles dans l'ARNr 23S sont associées à la résistance au linézolide. Des cas d'Enterococcus faecium résistant à la vancomycine devenant résistant au linézolide au cours de son utilisation clinique ont été signalés. Il y a des rapports de Staphylococcus aureus (résistant à la méthicilline) développant une résistance au linézolide pendant l'utilisation clinique. La résistance au linézolide chez ces organismes est associée à une mutation ponctuelle de l'ARNr 23S (substitution de la thymine à la guanine en position 2576) de l'organisme. Les organismes résistants aux oxazolidinones via des mutations dans les gènes chromosomiques codant pour l'ARNr 23S ou les protéines ribosomiques (L3 et L4) présentent généralement une résistance croisée au linézolide. Une résistance au linézolide chez les staphylocoques médiée par l'enzyme méthyltransférase a également été rapportée. Cette résistance est médiée par le gène cfr (chloramphénicol-florfénicol) situé sur un plasmide transférable entre staphylocoques.
Interaction avec d'autres médicaments antimicrobiens
Des études in vitro ont démontré une additivité ou une indifférence entre le linézolide et la vancomycine, la gentamicine, la rifampicine, l'imipénème-cilastatine, l'aztréonam, l'ampicilline ou la streptomycine.
Le linézolide s'est avéré actif contre la plupart des isolats des micro-organismes suivants, à la fois in vitro et dans les infections cliniques [voir LES INDICATIONS ].
Bactéries Gram-positives
Enterococcus faecium (isolats résistants à la vancomycine uniquement) Staphylococcus aureus (y compris les isolats résistants à la méthicilline) Streptococcus agalactiae Streptococcus pneumoniae Streptococcus pyogenes
Les données in vitro suivantes sont disponibles, mais leur signification clinique est inconnue. Plus de 90 % des bactéries suivantes présentent une CMI in vitro inférieure ou égale au point critique sensible au linézolide pour les organismes de genre similaire. L'innocuité et l'efficacité du linézolide dans le traitement des infections cliniques dues à ces bactéries n'ont pas été établies dans des essais cliniques adéquats et bien contrôlés.
Bactéries Gram-positives
Enterococcus faecalis (y compris les isolats résistants à la vancomycine) Enterococcus faecium (isolats sensibles à la vancomycine) Staphylococcus epidermidis (y compris les isolats résistants à la méthicilline) Staphylococcus haemolyticus Streptocoques du groupe Viridans
Bactéries Gram-négatives
Pasteurella multocida
Test de sensibilité
Pour des informations spécifiques concernant les critères d'interprétation des tests de sensibilité et les méthodes de test associées et les normes de contrôle qualité reconnues par la FDA pour ce médicament, veuillez consulter : https://www.fda.gov/STIC.
Toxicologie animale et/ou pharmacologie
Les organes cibles de la toxicité du linézolide étaient similaires chez les rats et les chiens juvéniles et adultes. Une myélosuppression dépendante de la dose et du temps, mise en évidence par une hypocellularité de la moelle osseuse/une diminution de l'hématopoïèse, une diminution de l'hématopoïèse extramédullaire dans la rate et le foie et une diminution des taux d'érythrocytes, de leucocytes et de plaquettes circulants, a été observée dans des études animales. Une déplétion lymphoïde s'est produite dans le thymus, les ganglions lymphatiques et la rate. Généralement, les résultats lymphoïdes étaient associés à l'anorexie, à la perte de poids et à la suppression du gain de poids corporel, ce qui peut avoir contribué aux effets observés.
Chez des rats ayant reçu du linézolide par voie orale pendant 6 mois, une dégénérescence axonale minime à légère irréversible des nerfs sciatiques a été observée à 80 mg/kg/jour; une dégénérescence minime du nerf sciatique a également été observée chez 1 mâle à ce niveau de dose lors d'une autopsie intermédiaire de 3 mois. Une évaluation morphologique sensible des tissus fixés par perfusion a été menée pour étudier les preuves de dégénérescence du nerf optique. Une dégénérescence minimale à modérée du nerf optique était évidente chez 2 rats mâles après 6 mois d'administration, mais la relation directe avec le médicament était équivoque en raison de la nature aiguë de la découverte et de sa distribution asymétrique. La dégénérescence nerveuse observée était comparable au microscope à la dégénérescence spontanée unilatérale du nerf optique signalée chez les rats vieillissants et peut être une exacerbation d'un changement de fond commun.
Ces effets ont été observés à des niveaux d'exposition comparables à ceux observés chez certains sujets humains. Les effets hématopoïétiques et lymphoïdes étaient réversibles, bien que dans certaines études, l'inversion ait été incomplète pendant la durée de la période de récupération.
Etudes cliniques
Adultes
Pneumonie nosocomiale
Des patients adultes atteints de pneumonie nosocomiale cliniquement et radiologiquement documentée ont été inclus dans un essai randomisé, multicentrique, en double aveugle. Les patients ont été traités pendant 7 à 21 jours. Un groupe a reçu ZYVOX IV Injection 600 mg toutes les 12 heures et l'autre groupe a reçu 1 g de vancomycine toutes les 12 heures par voie intraveineuse. Les deux groupes ont reçu de l'aztréonam concomitant (1 à 2 g toutes les 8 heures par voie intraveineuse), qui pouvait être poursuivi si cliniquement indiqué. Il y avait 203 patients traités au linézolide et 193 patients traités à la vancomycine inclus dans l'étude. Cent vingt-deux (60 %) patients traités par le linézolide et 103 (53 %) patients traités par la vancomycine étaient cliniquement évaluables. Les taux de guérison chez les patients cliniquement évaluables étaient de 57 % pour les patients traités au linézolide et de 60 % pour les patients traités à la vancomycine. Les taux de guérison chez les patients cliniquement évaluables atteints de pneumonie sous ventilation assistée étaient de 47 % pour les patients traités au linézolide et de 40 % pour les patients traités à la vancomycine. Une analyse en intention de traiter modifiée (MITT) de 94 patients traités au linézolide et de 83 patients traités à la vancomycine comprenait des sujets chez lesquels un agent pathogène avait été isolé avant le traitement. Les taux de guérison dans l'analyse MITT étaient de 57 % chez les patients traités au linézolide et de 46 % chez les patients traités à la vancomycine. Les taux de guérison par agent pathogène pour les patients microbiologiquement évaluables sont présentés dans le tableau 12.
Infections compliquées de la peau et des structures cutanées
Des patients adultes atteints d'infections compliquées de la peau et des structures cutanées cliniquement documentées ont été recrutés dans un essai randomisé, multicentrique, en double aveugle et double factice comparant les médicaments à l'étude administrés par voie intraveineuse suivis de médicaments administrés par voie orale pendant un total de 10 à 21 jours de traitement. Un groupe de patients a reçu ZYVOX IV Injection 600 mg toutes les 12 heures suivi de ZYVOX 600 mg Comprimés 600 mg toutes les 12 heures ; l'autre groupe a reçu de l'oxacilline 2 g toutes les 6 heures par voie intraveineuse suivie de dicloxacilline 500 mg toutes les 6 heures par voie orale. Les patients pouvaient recevoir de l'aztréonam en concomitance si cela était cliniquement indiqué. Il y avait 400 patients traités au linézolide et 419 patients traités à l'oxacilline inclus dans l'étude. Deux cent quarante-cinq (61 %) patients traités par linézolide et 242 (58 %) patients traités par oxacilline étaient cliniquement évaluables. Les taux de guérison chez les patients cliniquement évaluables étaient de 90 % chez les patients traités par le linézolide et de 85 % chez les patients traités par l'oxacilline. Une analyse en intention de traiter modifiée (MITT) de 316 patients traités au linézolide et de 313 patients traités à l'oxacilline a inclus des sujets qui remplissaient tous les critères d'admission à l'étude. Les taux de guérison dans l'analyse MITT étaient de 86 % chez les patients traités par le linézolide et de 82 % chez les patients traités par l'oxacilline. Les taux de guérison par agent pathogène pour les patients microbiologiquement évaluables sont présentés dans le tableau 13.
Une étude distincte a fourni une expérience supplémentaire avec l'utilisation de ZYVOX dans le traitement des infections à Staphylococcus aureus résistant à la méthicilline (SARM). Il s'agissait d'un essai randomisé ouvert chez des patients adultes hospitalisés présentant une infection à SARM documentée ou suspectée.
Un groupe de patients a reçu ZYVOX IV Injection 600 mg toutes les 12 heures suivi de ZYVOX Tablets 600 mg toutes les 12 heures. L'autre groupe de patients a reçu de la vancomycine 1 g toutes les 12 heures par voie intraveineuse. Les deux groupes ont été traités pendant 7 à 28 jours et pouvaient recevoir de l'aztréonam ou de la gentamicine en concomitance si cela était cliniquement indiqué. Les taux de guérison chez les patients microbiologiquement évaluables atteints d'infection de la peau et des structures cutanées à SARM étaient de 26/33 (79 %) pour les patients traités au linézolide et de 24/33 (73 %) pour les patients traités à la vancomycine.
Infections du pied diabétique
Des patients diabétiques adultes présentant des infections compliquées de la peau et des structures cutanées (« infections du pied diabétique ») ont été inclus dans un essai randomisé (ratio 2:1), multicentrique et ouvert comparant les médicaments à l'étude administrés par voie intraveineuse ou orale pendant un total de 14 à 28 jours de traitement. Un groupe de patients a reçu ZYVOX 600 mg toutes les 12 heures par voie intraveineuse ou orale ; l'autre groupe a reçu de l'ampicilline/sulbactam 1,5 à 3 g par voie intraveineuse ou de l'amoxicilline/acide clavulanique 500 à 875 mg toutes les 8 à 12 heures par voie orale. Dans les pays où l'ampicilline/sulbactam n'est pas commercialisé, l'amoxicilline/acide clavulanique 500 mg à 2 g toutes les 6 heures a été utilisé pour le schéma intraveineux. Les patients du groupe comparateur pouvaient également être traités par la vancomycine 1 g toutes les 12 heures par voie intraveineuse si le SARM était isolé de l'infection du pied. Les patients de l'un ou l'autre des groupes de traitement qui avaient des bacilles à Gram négatif isolés du site de l'infection pouvaient également recevoir de l'aztréonam 1 à 2 g toutes les 8 à 12 heures par voie intraveineuse. Tous les patients étaient éligibles pour recevoir des méthodes de traitement d'appoint appropriées, telles que le débridement et la décharge, comme cela est généralement requis dans le traitement des infections du pied diabétique, et la plupart des patients ont reçu ces traitements. Il y avait 241 patients traités par le linézolide et 120 patients traités par un comparateur dans la population de l'étude en intention de traiter (ITT). Deux cent douze (86 %) patients traités par le linézolide et 105 (85 %) patients traités par un comparateur étaient cliniquement évaluables. Dans la population ITT, les taux de guérison étaient de 68,5 % (165/241) chez les patients traités par le linézolide et de 64 % (77/120) chez les patients traités par un comparateur, ceux dont les résultats étaient indéterminés et manquants étaient considérés comme des échecs. Les taux de guérison chez les patients cliniquement évaluables (à l'exclusion de ceux dont les résultats étaient indéterminés et manquants) étaient de 83 % (159/192) et de 73 % (74/101) chez les patients traités par le linézolide et le comparateur, respectivement. Une analyse post-hoc critique s'est concentrée sur 121 patients traités par le linézolide et 60 patients traités par un comparateur qui présentaient un agent pathogène à Gram positif isolé du site de l'infection ou du sang, qui présentaient moins de signes d'ostéomyélite sous-jacente que la population globale de l'étude, et qui n'ont pas reçu d'antimicrobiens interdits. Sur la base de cette analyse, les taux de guérison étaient de 71 % (86/121) chez les patients traités par le linézolide et de 63 % (38/60) chez les patients traités par le comparateur. Aucune des analyses ci-dessus n'a été ajustée pour l'utilisation de thérapies complémentaires. Les taux de guérison par agent pathogène pour les patients microbiologiquement évaluables sont présentés dans le tableau 14.
Infections à entérocoques résistantes à la vancomycine
Des patients adultes présentant une infection à entérocoque résistante à la vancomycine documentée ou suspectée ont été recrutés dans un essai randomisé, multicentrique, en double aveugle comparant une dose élevée de ZYVOX (600 mg) à une faible dose de ZYVOX (200 mg) administrée toutes les 12 heures soit par voie intraveineuse (IV) ou par voie orale pendant 7 à 28 jours. Les patients pouvaient recevoir de l'aztréonam ou des aminoglycosides en concomitance. Il y avait 79 patients randomisés pour le linézolide à forte dose et 66 pour le linézolide à faible dose. La population en intention de traiter (ITT) avec une infection à entérocoque résistante à la vancomycine documentée au départ comprenait 65 patients dans le bras à dose élevée et 52 dans le bras à faible dose.
Les taux de guérison pour la population ITT avec une infection à entérocoque résistante à la vancomycine documentée au départ sont présentés dans le tableau 15 par source d'infection. Ces taux de guérison n'incluent pas les patients dont les résultats sont manquants ou indéterminés. Le taux de guérison était plus élevé dans le bras à forte dose que dans le bras à faible dose, bien que la différence ne soit pas statistiquement significative au niveau de 0,05.
Patients pédiatriques
Infections dues à des bactéries Gram-positives
Une étude d'innocuité et d'efficacité a fourni une expérience sur l'utilisation de ZYVOX 600 mg chez les patients pédiatriques pour le traitement de la pneumonie nosocomiale, des infections compliquées de la peau et des structures cutanées et d'autres infections dues à des pathogènes bactériens à Gram positif, y compris Staphylococcus aureus résistant et sensible à la méthicilline et Enterococcus faecium résistant à la vancomycine. Des patients pédiatriques âgés de la naissance à 11 ans avec des infections causées par les bactéries Gram-positives documentées ou suspectées ont été inscrits dans un essai randomisé, ouvert et contrôlé par comparateur. Un groupe de patients a reçu ZYVOX 600 mg IV Injection 10 mg/kg toutes les 8 heures suivi de ZYVOX 600 mg pour suspension orale 10 mg/kg toutes les 8 heures. Un deuxième groupe a reçu de la vancomycine 10 à 15 mg/kg par voie intraveineuse toutes les 6 à 24 heures, selon l'âge et la clairance rénale. Les patients qui avaient des infections à VRE confirmées ont été placés dans un troisième bras de l'étude et ont reçu ZYVOX 10 mg/kg toutes les 8 heures par voie intraveineuse et/ou orale. Tous les patients ont été traités pendant un total de 10 à 28 jours et ont pu recevoir en concomitance des médicaments antibactériens à Gram négatif si cela était cliniquement indiqué. Dans la population en intention de traiter (ITT), il y avait 206 patients randomisés pour le linézolide et 102 patients randomisés pour la vancomycine. Les taux de guérison pour les patients ITT, MITT et cliniquement évaluables sont présentés dans le tableau 16. Une fois l'étude terminée, 13 patients supplémentaires âgés de 4 jours à 16 ans ont été inclus dans une extension en ouvert du bras VRE du étude. Le tableau 17 présente les taux de guérison clinique par agent pathogène pour les patients microbiologiquement évaluables, y compris les patients microbiologiquement évaluables atteints d'Enterococcus faecium résistant à la vancomycine à partir de l'extension de cette étude.
INFORMATIONS PATIENTS
Instructions d'administration importantes
Avisez les patients que ZYVOX peut être pris avec ou sans nourriture.
Neuropathie périphérique et optique
Conseillez aux patients d'informer leur médecin s'ils ressentent des changements dans leur vision pendant qu'ils prennent ZYVOX [voir AVERTISSEMENTS ET PRECAUTIONS ].
Syndrome sérotoninergique
Conseillez aux patients d'informer leur médecin s'ils prennent des agents sérotoninergiques, y compris des inhibiteurs de la recapture de la sérotonine ou d'autres antidépresseurs et opioïdes [voir AVERTISSEMENTS ET PRECAUTIONS ].
Interactions potentielles produisant une élévation de la tension artérielle
- Conseillez aux patients d'informer leur médecin s'ils ont des antécédents d'hypertension.
- Conseillez aux patients d'éviter de grandes quantités d'aliments ou de boissons à forte teneur en tyramine pendant la prise de ZYVOX. Les aliments riches en tyramine comprennent ceux qui peuvent avoir subi des modifications protéiques par vieillissement, fermentation, marinage ou fumage pour améliorer la saveur, comme les fromages vieillis, les viandes fermentées ou séchées à l'air, la choucroute, la sauce soja, les bières pression et les vins rouges. La teneur en tyramine de tout aliment riche en protéines peut être augmentée s'il est conservé pendant de longues périodes ou mal réfrigéré.
- Conseillez aux patients d'informer leur médecin s'ils prennent des médicaments contenant du chlorhydrate de pseudoéphédrine ou du chlorhydrate de phénylpropanolamine, tels que des remèdes contre le rhume et des décongestionnants [voir AVERTISSEMENTS ET PRECAUTIONS ].
Acidose lactique
Conseillez aux patients d'informer leur médecin s'ils présentent des épisodes répétés de nausées ou de vomissements pendant le traitement par ZYVOX [voir AVERTISSEMENTS ET PRECAUTIONS ].
Convulsions
Conseillez aux patients d'informer leur médecin s'ils ont des antécédents de crises d'épilepsie ou de convulsions [voir AVERTISSEMENTS ET PRECAUTIONS ].
Hypoglycémie
Conseillez aux patients d'informer leur médecin s'ils souffrent de diabète sucré. Des réactions hypoglycémiques, telles qu'une diaphorèse et des tremblements, ainsi que des mesures de faible glycémie peuvent survenir lors d'un traitement par le linézolide. Si de telles réactions se produisent, les patients doivent contacter un médecin ou un autre professionnel de la santé pour un traitement approprié [voir AVERTISSEMENTS ET PRECAUTIONS ].
Hyponatrémie Et/ou SIADH
Conseillez aux patients à risque d'hyponatrémie d'informer leur médecin s'ils présentent des signes et des symptômes d'hyponatrémie et/ou de SIADH, y compris confusion, somnolence, faiblesse généralisée et détresse respiratoire [voir AVERTISSEMENTS ET PRECAUTIONS ].
Phénylcétonurie
Avisez les patients atteints de phénylcétonurie (PCU) que chaque 5 mL de 100 mg/5 mL de ZYVOX pour suspension orale contient 20 mg de phénylalanine. Les autres formulations de ZYVOX 600 mg ne contiennent pas de phénylalanine. La phénylalanine peut être nocive pour les patients atteints de phénylcétonurie. Contactez votre médecin ou votre pharmacien lorsqu'il vous est prescrit avec ZYVOX 600 mg pour suspension buvable [voir AVERTISSEMENTS ET PRECAUTIONS ].
Résistance antibactérienne
Les patients doivent être informés que les médicaments antibactériens, y compris ZYVOX, ne doivent être utilisés que pour traiter les infections bactériennes. Ils ne traitent pas les infections virales (par exemple, le rhume). Lorsque ZYVOX 600 mg est prescrit pour traiter une infection bactérienne, les patients doivent être informés que bien qu'il soit courant de se sentir mieux au début du traitement, le médicament doit être pris exactement comme indiqué. Sauter des doses ou ne pas terminer le traitement complet peut diminuer l'efficacité du traitement immédiat et augmenter la probabilité que les bactéries développent une résistance et ne puissent plus être traitées par ZYVOX ou d'autres médicaments antibactériens à l'avenir.
Diarrhée
La diarrhée est un problème courant causé par les médicaments antibactériens, qui se termine généralement lorsque le médicament antibactérien est arrêté. Parfois, après le début du traitement avec des médicaments antibactériens, les patients peuvent développer des selles liquides et sanglantes (avec ou sans crampes d'estomac et fièvre) même deux mois ou plus après avoir pris la dernière dose du médicament antibactérien. Si cela se produit, les patients doivent contacter leur médecin dès que possible [voir AVERTISSEMENTS ET PRECAUTIONS ].
Infertilité
Informez les patients de sexe masculin que ZYVOX peut altérer de manière réversible la fertilité [voir Utilisation dans des populations spécifiques ].
L'étiquetage de ce produit peut avoir été mis à jour. Pour les informations de prescription les plus récentes, veuillez visiter www.pfizer.com.