Omnicef 300mg Cefdinir Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce qu'Omnicef et comment est-il utilisé ?
Omnicef (cefdinir) est un antibiotique céphalosporine utilisé pour traiter de nombreux types d'infections causées par des bactéries. Le nom de marque Omnicef est abandonné aux États-Unis. Omnicef est disponible sous forme générique.
Quels sont les effets secondaires d'Omnicef ?
Les effets secondaires courants d'Omnicef 300 mg incluent :
- diarrhée,
- nausée,
- vomissement,
- Douleur d'estomac,
- indigestion,
- mal de tête,
- vertiges,
- érythème fessier chez un nourrisson prenant du cefdinir liquide,
- démangeaisons ou
- démangeaison de la peau,
Noter: Cette liste peut ne pas inclure tous les effets secondaires possibles.
Pour réduire le développement de bactéries résistantes aux médicaments et maintenir l'efficacité d'OMNICEF 300 mg et d'autres médicaments antibactériens, OMNICEF doit être utilisé uniquement pour traiter ou prévenir les infections dont il est prouvé ou fortement suspecté qu'elles sont causées par des bactéries.
LA DESCRIPTION
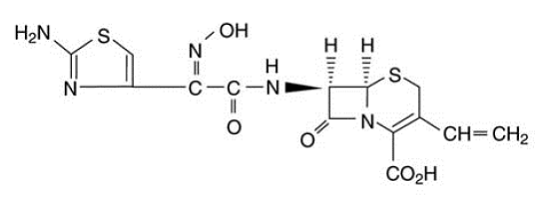
Les capsules OMNICEF (cefdinir) et OMNICEF (cefdinir) pour suspension buvable contiennent l'ingrédient actif cefdinir, une céphalosporine semi-synthétique à spectre étendu, pour administration orale. Chimiquement, le cefdinir est [6R-[6α, 7β (Z)]]-7-[[(2-amino-4-thiazolyl)(hydroxyimino)acétyl]amino]3-éthényl-8-oxo-5-thia-1 Acide -azabicyclo[4.2.0]oct-2-ène-2-carboxylique. Le cefdinir est un solide blanc à jaune légèrement brunâtre. Il est légèrement soluble dans l'acide chlorhydrique dilué et peu soluble dans le tampon phosphate 0,1 M pH 7,0. La formule empirique est C14H13N5O5S2 et le poids moléculaire est de 395,42. Cefdinir a la formule structurelle indiquée ci-dessous :
Les gélules OMNICEF 300 mg contiennent 300 mg de cefdinir et les ingrédients inactifs suivants : carboxyméthylcellulose calcique, NF ; stéarate de polyoxyle 40, NF; et stéarate de magnésium, NF. Les enveloppes des gélules contiennent du FD&C Blue #1 ; FD&C rouge n° 40 ; D&C Rouge #28; dioxyde de titane, NF; gélatine, NF; dioxyde de silicium, NF; et le laurylsulfate de sodium, NF.
OMNICEF 300 mg pour suspension buvable, après reconstitution, contient 125 mg de cefdinir pour 5 mL ou 250 mg de cefdinir pour 5 mL et les ingrédients inactifs suivants : saccharose, NF ; acide citrique, USP; citrate de sodium, USP; benzoate de sodium, NF; gomme xanthane, NF; gomme de guar, NF; arômes artificiels de fraise et de crème; dioxyde de silicium, NF; et stéarate de magnésium, NF.
LES INDICATIONS
Pour réduire le développement de bactéries résistantes aux médicaments et maintenir l'efficacité d'OMNICEF 300 mg et d'autres médicaments antibactériens, OMNICEF 300 mg doit être utilisé uniquement pour traiter ou prévenir les infections dont il est prouvé ou fortement suspecté qu'elles sont causées par des bactéries sensibles. Lorsque des informations sur la culture et la sensibilité sont disponibles, elles doivent être prises en compte lors de la sélection ou de la modification du traitement antibactérien. En l'absence de telles données, l'épidémiologie locale et les schémas de sensibilité peuvent contribuer à la sélection empirique du traitement.
Les capsules OMNICEF (cefdinir) et OMNICEF (cefdinir) pour suspension buvable sont indiquées pour le traitement des patients atteints d'infections légères à modérées causées par des souches sensibles des micro-organismes désignés dans les conditions énumérées ci-dessous.
Adultes et adolescents
Pneumonie communautaire
causée par Haemophilus influenzae (y compris les souches productrices de β-lactamases), Haemophilus parainfluenzae (y compris les souches productrices de β-lactamases), Streptococcus pneumoniae (souches sensibles à la pénicilline uniquement) et Moraxella catarrhalis (y compris les souches productrices de β-lactamases) (voir Etudes cliniques ).
Exacerbations aiguës de la bronchite chronique
causée par Haemophilus influenzae (y compris les souches productrices de β-lactamases), Haemophilus parainfluenzae (y compris les souches productrices de β-lactamases), Streptococcus pneumoniae (souches sensibles à la pénicilline uniquement) et Moraxella catarrhalis (y compris les souches productrices de β-lactamases).
Sinusite maxillaire aiguë
causée par Haemophilus influenzae (y compris les souches productrices de β-lactamase), Streptococcus pneumoniae (souches sensibles à la pénicilline uniquement) et Moraxella catarrhalis (y compris les souches productrices de βlactamase).
REMARQUE: Pour plus d'informations sur l'utilisation chez les patients pédiatriques, voir Utilisation pédiatrique et DOSAGE ET ADMINISTRATION .
Pharyngite/amygdalite
causée par Streptococcus pyogenes (voir Etudes cliniques ).
REMARQUE: Le cefdinir est efficace dans l'éradication de S. pyogenes de l'oropharynx. Le cefdinir n'a cependant pas été étudié pour la prévention du rhumatisme articulaire aigu consécutif à une pharyngite/amygdalite à S. pyogenes. Seule la pénicilline intramusculaire s'est avérée efficace pour la prévention du rhumatisme articulaire aigu.
Infections non compliquées de la peau et des structures cutanées
causée par Staphylococcus aureus (y compris les souches productrices de β-lactamase) et Streptococcus pyogenes.
Patients pédiatriques
Otite moyenne bactérienne aiguë causée par Haemophilus influenzae (y compris les souches productrices de β-lactamases), Streptococcus pneumoniae (souches sensibles à la pénicilline uniquement) et Moraxella catarrhalis (y compris les souches productrices de β-lactamases).
Pharyngite/amygdalite
causée par Streptococcus pyogenes (voir Etudes cliniques ).
REMARQUE: Le cefdinir est efficace dans l'éradication de S. pyogenes de l'oropharynx. Le cefdinir n'a cependant pas été étudié pour la prévention du rhumatisme articulaire aigu consécutif à une pharyngite/amygdalite à S. pyogenes. Seule la pénicilline intramusculaire s'est avérée efficace pour la prévention du rhumatisme articulaire aigu.
Infections non compliquées de la peau et des structures cutanées
causée par Staphylococcus aureus (y compris les souches productrices de β-lactamase) et Streptococcus pyogenes.
DOSAGE ET ADMINISTRATION
(voir INDICATIONS ET USAGE pour Agents pathogènes indiqués )
Gélules
La posologie et la durée de traitement recommandées pour les infections chez les adultes et les adolescents sont décrites dans le tableau suivant ; la dose quotidienne totale pour toutes les infections est de 600 mg. L'administration une fois par jour pendant 10 jours est aussi efficace que l'administration BID. L'administration uniquotidienne n'a pas été étudiée dans la pneumonie ou les infections cutanées; par conséquent, les capsules OMNICEF doivent être administrées deux fois par jour dans ces infections. Les gélules OMNICEF peuvent être prises sans tenir compte des repas.
Adultes et adolescents (13 ans et plus)
PoudrePour suspension orale
La posologie et la durée de traitement recommandées pour les infections chez les patients pédiatriques sont décrites dans le tableau suivant ; la dose quotidienne totale pour toutes les infections est de 14 mg/kg, jusqu'à une dose maximale de 600 mg par jour. L'administration une fois par jour pendant 10 jours est aussi efficace que l'administration BID. L'administration uniquotidienne n'a pas été étudiée dans les infections cutanées; par conséquent, OMNICEF 300 mg pour suspension orale doit être administré deux fois par jour dans cette infection. OMNICEF pour suspension orale peut être administré sans tenir compte des repas.
Patients pédiatriques (âgés de 6 mois à 12 ans)
TABLEAU DE POSOLOGIE PÉDIATRIQUE D'OMNICEF POUR SUSPENSION BUVABLE
Patients insuffisants rénaux
Chez les patients adultes dont la clairance de la créatinine est
La clairance de la créatinine est difficile à mesurer en ambulatoire. Cependant, la formule suivante peut être utilisée pour estimer la clairance de la créatinine (CLcr) chez les patients adultes. Pour que les estimations soient valides, les taux de créatinine sérique doivent refléter les niveaux de la fonction rénale à l'état d'équilibre.
où la clairance de la créatinine est en mL/min, l'âge est en années, le poids est en kilogrammes et la créatinine sérique est en mg/dL.4
La formule suivante peut être utilisée pour estimer la clairance de la créatinine chez les patients pédiatriques :
CLcr = K × longueur ou taille corporelle/créatinine sérique
où K = 0,55 pour les patients pédiatriques de plus de 1 an5 et 0,45 pour les nourrissons (jusqu'à 1 an)6.
Dans l'équation ci-dessus, la clairance de la créatinine est en mL/min/1,73 m², la longueur ou la taille du corps est en centimètres et la créatinine sérique est en mg/dL.
Pour les patients pédiatriques avec une clairance de la créatinine
Patients sous hémodialyse
L'hémodialyse élimine le cefdinir du corps. Chez les patients maintenus sous hémodialyse chronique, le schéma posologique initial recommandé est une dose de 300 mg ou 7 mg/kg tous les deux jours.
A la fin de chaque séance d'hémodialyse, 300 mg (ou 7 mg/kg) doivent être administrés. Les doses suivantes (300 mg ou 7 mg/kg) sont ensuite administrées tous les deux jours.
Indications pour mélanger Omnicef 300 mg pour suspension buvable
Après mélange, la suspension peut être conservée à température ambiante (25°C/77°F). Le récipient doit être maintenu hermétiquement fermé et la suspension doit être bien agitée avant chaque administration. La suspension peut être utilisée pendant 10 jours, après quoi toute portion inutilisée doit être jetée.
COMMENT FOURNIE
Gélules OMNICEF , contenant 300 mg de cefdinir, sous forme de gélules lavande et turquoise portant le nom du produit, sont disponibles comme suit :
60 gélules/bouteille CDN 0074-3769-60 Boîte OMNI-PAC™ de 3 plaquettes alvéolées de 10 capsules, d'une durée de 5 jours et d'une unité d'utilisation CDN 0074-3769-30
OMNICEF 300 mg pour suspension buvable est une formulation en poudre de couleur crème qui, lorsqu'elle est reconstituée selon les instructions, contient 125 mg de cefdinir/5 mL ou 250 mg de cefdinir/5 mL. Les suspensions reconstituées ont une couleur crème et un goût de fraise. La poudre est disponible comme suit :
125 mg/5 mL
Flacons de 60 ml CDN 0074-3771-60 Bouteilles de 100 ml CDN 0074-3771-13 250 mg/5 mL Flacons de 60 mL CDN 0074-6151-60 Bouteilles de 100 ml CDN 0074-6151-13
Conserver les gélules et la poudre non suspendue à 25°C (77°F); les excursions permises à 15°-30°C (59°-86°F) [voient USP la Température de Pièce Contrôlée]. Une fois reconstituée, la suspension buvable peut être conservée à température ambiante contrôlée pendant 10 jours.
Fabriqué par : CEPH International Corporation Carolina, Porto Rico 00986. Pour : AbbVie Inc., North Chicago, IL 60064, États-Unis, sous licence de : Astellas Pharma Inc. Tokyo, Japon. Révisé en novembre 2015
EFFETS SECONDAIRES
Événements indésirables
Essais cliniques - Capsules OMNICEF (Patients adultes et adolescents)
Dans les essais cliniques, 5093 patients adultes et adolescents (3841 américains et 1252 non américains) ont été traités avec la dose recommandée de gélules de cefdinir (600 mg/jour). La plupart des événements indésirables étaient légers et spontanément résolutifs. Aucun décès ou incapacité permanente n'a été attribué au cefdinir. Cent quarante-sept des 5093 (3%) patients ont arrêté le traitement en raison d'effets indésirables que les investigateurs pensaient être possiblement, probablement ou définitivement associés au traitement par le cefdinir. Les arrêts étaient principalement dus à des troubles gastro-intestinaux, généralement des diarrhées ou des nausées. Dix-neuf des 5093 (0,4 %) patients ont été arrêtés en raison d'une éruption cutanée liée à l'administration de cefdinir.
Aux États-Unis, les effets indésirables suivants ont été considérés par les chercheurs comme étant possiblement, probablement ou définitivement liés aux gélules de cefdinir dans des essais cliniques à doses multiples (N = 3 841 patients traités par cefdinir) :
ÉVÉNEMENTS INDÉSIRABLES ASSOCIÉS AUX CAPSULES DE CEFDINIR ESSAIS AUX ÉTATS-UNIS CHEZ DES PATIENTS ADULTES ET ADOLESCENTS (N = 3841)a
Les changements de valeur de laboratoire suivants, potentiellement significatifs sur le plan clinique, indépendamment de la relation avec le traitement par le cefdinir, ont été observés au cours des essais cliniques menés aux États-Unis :
MODIFICATIONS DES VALEURS DE LABORATOIRE OBSERVÉES AVEC CEFDINIR CAPSULES ESSAIS US CHEZ DES PATIENTS ADULTES ET ADOLESCENTS (N = 3841)
Essais cliniques - OMNICEF pour suspension orale (patients pédiatriques)
Dans les essais cliniques, 2289 patients pédiatriques (1783 américains et 506 non américains) ont été traités avec la dose recommandée de suspension de cefdinir (14 mg/kg/jour). La plupart des événements indésirables étaient légers et spontanément résolutifs. Aucun décès ou incapacité permanente n'a été attribué au cefdinir. Quarante des 2289 (2%) patients ont arrêté le traitement en raison d'événements indésirables considérés par les investigateurs comme étant possiblement, probablement ou définitivement associés au traitement par le cefdinir. Les arrêts étaient principalement dus à des troubles gastro-intestinaux, généralement des diarrhées. Cinq des 2289 (0,2 %) patients ont été arrêtés en raison d'une éruption cutanée liée à l'administration de cefdinir.
Aux États-Unis, les effets indésirables suivants ont été considérés par les investigateurs comme étant possiblement, probablement ou définitivement liés à la suspension de cefdinir dans des essais cliniques à doses multiples (N = 1 783 patients non traités au cefdinir) :
EFFETS INDÉSIRABLES ASSOCIÉS AUX ESSAIS DE SUSPENSION DU CEFDINIR CHEZ DES PATIENTS PÉDIATRIQUES (N = 1783)a
REMARQUE : Chez les patients traités au cefdinir et aux groupes témoins, les taux de diarrhée et d'éruptions cutanées étaient plus élevés chez les patients pédiatriques les plus jeunes. L'incidence de la diarrhée chez les patients âgés de ≤ 2 ans traités par cefdinir était de 17 % (95/557) contre 4 % (51/1226) chez ceux de > 2 ans. L'incidence des éruptions cutanées (principalement des érythèmes fessiers chez les patients plus jeunes) était de 8 % (43/557) chez les patients âgés de ≤ 2 ans contre 1 % (8/1226) chez ceux de > 2 ans.
Les changements de valeur de laboratoire suivants, potentiellement significatifs sur le plan clinique, indépendamment de la relation avec le traitement par le cefdinir, ont été observés au cours des essais cliniques menés aux États-Unis :
MODIFICATIONS DES VALEURS DE LABORATOIRE D'UNE SIGNIFICATION CLINIQUE POSSIBLE OBSERVÉES AVEC LES ESSAIS US DE SUSPENSION DU CEFDINIR CHEZ DES PATIENTS PÉDIATRIQUES (N = 1783)
Expérience post-commercialisation
Les effets indésirables suivants et les tests de laboratoire modifiés, quelle que soit leur relation avec le cefdinir, ont été rapportés au cours d'une vaste expérience post-commercialisation, à commencer par l'approbation au Japon en 1991 : choc, anaphylaxie avec de rares cas de décès, œdème facial et laryngé, sensation d'étouffement, réactions de type maladie sérique, conjonctivite, stomatite, syndrome de Stevens-Johnson, nécrolyse épidermique toxique, dermatite exfoliative, érythème polymorphe, érythème noueux, hépatite aiguë, cholestase, hépatite fulminante, insuffisance hépatique, ictère, augmentation de l'amylase, entérocolite aiguë, diarrhée sanglante, colite hémorragique, méléna, colite pseudomembraneuse, pancytopénie, granulocytopénie, leucopénie, thrombocytopénie, purpura thrombocytopénique idiopathique, anémie hémolytique, insuffisance respiratoire aiguë, crise d'asthme, pneumonie d'origine médicamenteuse, pneumonie à éosinophiles, pneumonie interstitielle idiopathique, fièvre, insuffisance rénale aiguë, néphropathie, tendance aux saignements, troubles de la coagulation troubles digestifs, coagulation intravasculaire disséminée, saignement gastro-intestinal supérieur, ulcère peptique, iléus, perte de conscience, vascularite allergique, interaction possible cefdinir-diclofénac, insuffisance cardiaque, douleur thoracique, infarctus du myocarde, hypertension, mouvements involontaires et rhabdomyolyse.
Événements indésirables de la classe des céphalosporines
Les événements indésirables suivants et les tests de laboratoire modifiés ont été signalés pour les antibiotiques de la classe des céphalosporines en général :
Réactions allergiques, anaphylaxie, syndrome de Stevens-Johnson, érythème polymorphe, nécrolyse épidermique toxique, dysfonctionnement rénal, néphropathie toxique, dysfonctionnement hépatique, y compris cholestase, anémie aplasique, anémie hémolytique, hémorragie, test de glucose urinaire faussement positif, neutropénie, pancytopénie et agranulocytose . Les symptômes de la colite pseudomembraneuse peuvent apparaître pendant ou après un traitement antibiotique (voir AVERTISSEMENTS ).
Plusieurs céphalosporines ont été impliquées dans le déclenchement des convulsions, en particulier chez les insuffisants rénaux lorsque la posologie n'était pas diminuée (voir DOSAGE ET ADMINISTRATION et SURDOSAGE ). Si des convulsions associées au traitement médicamenteux surviennent, le médicament doit être arrêté. Un traitement anticonvulsivant peut être administré si cliniquement indiqué.
INTERACTIONS MÉDICAMENTEUSES
Antiacides (contenant de l'aluminium ou du magnésium)
L'administration concomitante de capsules de cefdinir à 300 mg avec 30 mL de suspension de Maalox® TC réduit le taux (Cmax) et l'étendue (ASC) de l'absorption d'environ 40 %. Le temps nécessaire pour atteindre la Cmax est également prolongé d'une heure. Il n'y a pas d'effets significatifs sur la pharmacocinétique du cefdinir si l'antiacide est administré 2 heures avant ou 2 heures après le cefdinir. Si des antiacides sont nécessaires pendant le traitement par OMNICEF 300 mg, OMNICEF 300 mg doit être pris au moins 2 heures avant ou après l'antiacide.
Probénécide
Comme avec d'autres antibiotiques β-lactamines, le probénécide inhibe l'excrétion rénale du cefdinir, entraînant un doublement approximatif de l'ASC, une augmentation de 54 % des concentrations plasmatiques maximales de cefdinir et une prolongation de 50 % de la t½ d'élimination apparente.
Suppléments de fer et aliments enrichis en fer
L'administration concomitante de cefdinir avec un supplément de fer thérapeutique contenant 60 mg de fer élémentaire (sous forme de FeSO4) ou des vitamines complétées par 10 mg de fer élémentaire a réduit l'étendue de l'absorption de 80 % et 31 %, respectivement. Si des suppléments de fer sont nécessaires pendant le traitement par OMNICEF 300 mg, OMNICEF doit être pris au moins 2 heures avant ou après le supplément.
L'effet des aliments fortement enrichis en fer élémentaire (principalement des céréales pour petit-déjeuner enrichies en fer) sur l'absorption du cefdinir n'a pas été étudié.
L'administration concomitante de préparations pour nourrissons enrichies de fer (2,2 mg de fer élémentaire/6 oz) n'a aucun effet significatif sur la pharmacocinétique du cefdinir. Par conséquent, OMNICEF pour suspension orale peut être administré avec une préparation pour nourrissons enrichie de fer.
Des cas de selles rougeâtres ont été signalés chez des patients recevant du cefdinir. Dans de nombreux cas, les patients recevaient également des produits contenant du fer. La couleur rougeâtre est due à la formation d'un complexe non résorbable entre le cefdinir ou ses produits de dégradation et le fer dans le tractus gastro-intestinal.
Interactions médicament/test de laboratoire
Une réaction faussement positive pour les cétones dans l'urine peut se produire avec des tests utilisant du nitroprussiate, mais pas avec ceux utilisant du nitroferricyanure. L'administration de cefdinir peut entraîner une réaction faussement positive pour le glucose dans l'urine en utilisant Clinitest®, la solution de Benedict ou la solution de Fehling. Il est recommandé d'utiliser des tests de glycémie basés sur des réactions enzymatiques de glucose oxydase (tels que Clinistix® ou Tes-Tape®). Les céphalosporines sont connues pour induire occasionnellement un test de Coombs direct positif.
AVERTISSEMENTS
AVANT QUE LA THÉRAPIE AVEC OMNICEF (CEFDINIR) SOIT INSTITUÉE, UNE ENQUÊTE PRUDENTE DEVRAIT ÊTRE FAITE POUR DÉTERMINER SI LE PATIENT A EU DES RÉACTIONS D'HYPERSENSIBILITÉ PRÉCÉDENTES AU CEFDINIR, À D'AUTRES CÉPHALOSPORINES, À DES PÉNICILLINES OU À D'AUTRES MÉDICAMENTS. SI LE CEFDINIR DOIT ÊTRE ADMINISTRÉ À DES PATIENTS SENSIBLES À LA PÉNICILLINE, LA PRUDENCE DOIT ÊTRE EXERCÉE CAR L'HYPERSENSIBILITÉ CROISÉE PARMI LES ANTIBIOTIQUES β-LACTAM A ÉTÉ CLAIREMENT DOCUMENTÉE ET PEUT SURVENIR CHEZ JUSQU'À 10 % DES PATIENTS AYANT DES ANTÉCÉDENTS D'ALLERGIES À LA PÉNICILLINE. SI UNE RÉACTION ALLERGIQUE AU CEFDINIR SE PRODUIT, LE MÉDICAMENT DOIT ÊTRE ARRÊTÉ. LES RÉACTIONS D'HYPERSENSIBILITÉ AIGUË GRAVES PEUVENT NÉCESSIER UN TRAITEMENT À L'ÉPINÉPHRINE ET D'AUTRES MESURES D'URGENCE, Y COMPRIS L'OXYGÈNE, LES LIQUIDES INTRAVEINEUSES, LES ANTIHISTAMINES INTRAVEINEUSES, LES CORTICOSTÉROÏDES, LES AMINES PRESSEURS ET LA GESTION DES VOIES RESPIRATOIRES, SELON L'INDICATION CLINIQUE.
Des diarrhées associées à Clostridium difficile (DACD) ont été signalées avec l'utilisation de presque tous les agents antibactériens, y compris l'OMNICEF, et leur gravité peut varier d'une diarrhée légère à une colite mortelle. Le traitement avec des agents antibactériens altère la flore normale du côlon, entraînant une prolifération de C. difficile.
C. difficile produit les toxines A et B qui contribuent au développement de CDAD. Les souches de C. difficile productrices d'hypertoxines entraînent une morbidité et une mortalité accrues, car ces infections peuvent être réfractaires au traitement antimicrobien et nécessiter une colectomie. La DACD doit être envisagée chez tous les patients qui présentent une diarrhée suite à l'utilisation d'antibactériens. Des antécédents médicaux minutieux sont nécessaires car il a été rapporté que la DACD survient plus de deux mois après l'administration d'agents antibactériens.
Si la CDAD est suspectée ou confirmée, l'utilisation continue d'antibactériens non dirigés contre C. difficile peut devoir être interrompue. Une gestion appropriée des liquides et des électrolytes, une supplémentation en protéines, un traitement antibactérien de C. difficile et une évaluation chirurgicale doivent être institués selon les indications cliniques.
PRÉCAUTIONS
Général
Prescrire OMNICEF 300 mg en l'absence d'infection bactérienne avérée ou fortement suspectée ou d'indication prophylactique est peu susceptible d'apporter un bénéfice au patient et augmente le risque de développement de bactéries résistantes aux médicaments.
Comme avec d'autres antibiotiques à large spectre, un traitement prolongé peut entraîner l'émergence et la prolifération possibles d'organismes résistants. Une observation attentive du patient est essentielle. Si une surinfection survient pendant le traitement, un traitement alternatif approprié doit être administré.
Le cefdinir, comme les autres antimicrobiens à large spectre (antibiotiques), doit être prescrit avec prudence chez les personnes ayant des antécédents de colite.
Chez les patients présentant une insuffisance rénale transitoire ou persistante (clairance de la créatinine DOSAGE ET ADMINISTRATION ).
Carcinogenèse, mutagenèse, altération de la fertilité
Le potentiel carcinogène du cefdinir n'a pas été évalué. Aucun effet mutagène n'a été observé dans le test de mutation inverse bactérienne (Ames) ou le test de mutation ponctuelle au locus de l'hypoxanthine-guanine phosphoribosyltransférase (HGPRT) dans les cellules pulmonaires de hamster chinois V79. Aucun effet clastogène n'a été observé in vitro dans le test d'aberration chromosomique structurale dans les cellules pulmonaires de hamster chinois V79 ou in vivo dans le test du micronoyau dans la moelle osseuse de souris. Chez le rat, la fertilité et les performances de reproduction n'ont pas été affectées par le cefdinir à des doses orales allant jusqu'à 1000 mg/kg/jour (70 fois la dose humaine basée sur mg/kg/jour, 11 fois basée sur mg/m²/jour).
Grossesse
Effets tératogènes
Catégorie de grossesse B
Le cefdinir n'était pas tératogène chez le rat à des doses orales allant jusqu'à 1000 mg/kg/jour (70 fois la dose humaine basée sur mg/kg/jour, 11 fois basée sur mg/m²/jour) ou chez le lapin à des doses orales allant jusqu'à 10 mg/kg/jour (0,7 fois la dose humaine basée sur mg/kg/jour, 0,23 fois basée sur mg/m²/jour). Une toxicité maternelle (diminution du gain de poids corporel) a été observée chez les lapins à la dose maximale tolérée de 10 mg/kg/jour sans effets indésirables sur la progéniture. Une diminution du poids corporel est survenue chez les fœtus de rat à ≥ 100 mg/kg/jour et chez la progéniture de rat à ≥ 32 mg/kg/jour. Aucun effet n'a été observé sur les paramètres de reproduction maternelle ou la survie, le développement, le comportement ou la fonction de reproduction de la progéniture.
Il n'y a cependant pas d'études adéquates et bien contrôlées chez les femmes enceintes. Étant donné que les études sur la reproduction chez l'animal ne sont pas toujours prédictives de la réponse humaine, ce médicament ne doit être utilisé pendant la grossesse qu'en cas de nécessité absolue.
Travail et accouchement
Cefdinir n'a pas été étudié pour une utilisation pendant le travail et l'accouchement.
Mères allaitantes
Après l'administration de doses uniques de 600 mg, le cefdinir n'a pas été détecté dans le lait maternel humain.
Utilisation pédiatrique
La sécurité et l'efficacité chez les nouveau-nés et les nourrissons de moins de 6 mois n'ont pas été établies. L'utilisation de cefdinir pour le traitement de la sinusite maxillaire aiguë chez les patients pédiatriques (âgés de 6 mois à 12 ans) est étayée par des preuves issues d'études adéquates et bien contrôlées chez les adultes et les adolescents, la physiopathologie similaire de la sinusite aiguë chez les patients adultes et pédiatriques, et données pharmacocinétiques comparatives dans la population pédiatrique.
Utilisation gériatrique
L'efficacité est comparable chez les patients gériatriques et les jeunes adultes. Alors que le cefdinir a été bien toléré dans tous les groupes d'âge, dans les essais cliniques, les patients gériatriques ont présenté un taux plus faible d'événements indésirables, y compris la diarrhée, que les adultes plus jeunes. L'adaptation de la dose chez les patients âgés n'est pas nécessaire à moins que la fonction rénale ne soit nettement altérée (voir DOSAGE ET ADMINISTRATION ).
SURDOSAGE
Aucune information sur le surdosage de cefdinir chez l'homme n'est disponible. Dans les études de toxicité aiguë chez les rongeurs, une seule dose orale de 5600 mg/kg n'a produit aucun effet indésirable. Les signes et symptômes toxiques suite à un surdosage avec d'autres antibiotiques β-lactamines ont inclus des nausées, des vomissements, une détresse épigastrique, de la diarrhée et des convulsions. L'hémodialyse élimine le cefdinir du corps. Cela peut être utile en cas de réaction toxique grave due à un surdosage, en particulier si la fonction rénale est altérée.
CONTRE-INDICATIONS
OMNICEF (cefdinir) est contre-indiqué chez les patients présentant une allergie connue à la classe d'antibiotiques des céphalosporines.
PHARMACOLOGIE CLINIQUE
Pharmacocinétique et métabolisme des médicaments
Absorption
Biodisponibilité orale
Les concentrations plasmatiques maximales de cefdinir surviennent 2 à 4 heures après l'administration de la capsule ou de la suspension. Les concentrations plasmatiques de cefdinir augmentent avec la dose, mais les augmentations sont moins que proportionnelles à la dose de 300 mg (7 mg/kg) à 600 mg (14 mg/kg). Après administration de la suspension à des adultes sains, la biodisponibilité du cefdinir est de 120 % par rapport aux gélules. La biodisponibilité estimée des gélules de cefdinir est de 21 % après l'administration d'une dose de 300 mg de gélule et de 16 % après l'administration d'une dose de 600 mg de gélule. La biodisponibilité absolue estimée de la suspension de cefdinir est de 25 %. La suspension buvable de Cefdinir dosée à 250 mg/5 ml s'est avérée bioéquivalente à la dose de 125 mg/5 ml chez des adultes en bonne santé à jeun.
Effet de la nourriture
La Cmax et l'ASC du cefdinir des gélules sont réduites de 16 % et 10 %, respectivement, lorsqu'elles sont administrées avec un repas riche en graisses. Chez les adultes ayant reçu la suspension buvable à 250 mg/5 mL avec un repas riche en graisses, la Cmax et l'ASC du cefdinir sont réduites de 44 % et 33 %, respectivement. L'ampleur de ces réductions n'est probablement pas cliniquement significative car les études d'innocuité et d'efficacité de la suspension buvable chez les patients pédiatriques ont été menées sans tenir compte de l'apport alimentaire. Par conséquent, le cefdinir peut être pris sans égard à la nourriture.
Gélules de Cefdinir
Les concentrations plasmatiques de cefdinir et les valeurs des paramètres pharmacocinétiques après l'administration de doses orales uniques de 300 et 600 mg de cefdinir à des sujets adultes sont présentées dans le tableau suivant :
Valeurs moyennes (± ET) des paramètres pharmacocinétiques plasmatiques du cefdinir après administration de gélules à des sujets adultes
Cefdinir Suspension
Les concentrations plasmatiques de cefdinir et les valeurs des paramètres pharmacocinétiques après l'administration de doses orales uniques de 7 et 14 mg/kg de cefdinir à des sujets pédiatriques (âgés de 6 mois à 12 ans) sont présentées dans le tableau suivant :
Valeurs moyennes (± ET) des paramètres pharmacocinétiques plasmatiques du cefdinir après l'administration de la suspension à des sujets pédiatriques
Dosage multiple
Le cefdinir ne s'accumule pas dans le plasma après une administration une ou deux fois par jour chez des sujets ayant une fonction rénale normale.
Distribution
Le volume moyen de distribution (Vdarea) du cefdinir chez les sujets adultes est de 0,35 L/kg (± 0,29) ; chez les sujets pédiatriques (âgés de 6 mois à 12 ans), le cefdinir Vdarea est de 0,67 L/kg (± 0,38). Le cefdinir est lié à 60 % à 70 % aux protéines plasmatiques chez les sujets adultes et pédiatriques ; la liaison est indépendante de la concentration.
Peau Blister
Chez les sujets adultes, des concentrations maximales médianes (plage) de cefdinir dans le liquide vésiculaire de 0,65 (0,33-1,1) et 1,1 (0,49-1,9) μg/mL ont été observées 4 à 5 heures après l'administration de doses de 300 et 600 mg, respectivement. Les valeurs moyennes (± SD) de la Cmax et de l'ASC (0-∞) des cloques étaient de 48 % (± 13) et 91 % (± 18) des valeurs plasmatiques correspondantes.
Tissu d'amygdale
Chez les patients adultes subissant une amygdalectomie élective, les concentrations médianes respectives de cefdinir dans les tissus amygdaliens 4 heures après l'administration de doses uniques de 300 et 600 mg étaient de 0,25 (0,220,46) et 0,36 (0,22-0,80) μg/g. Les concentrations moyennes dans le tissu des amygdales étaient de 24 % (± 8) des concentrations plasmatiques correspondantes.
Tissu sinusal
Chez les patients adultes subissant une chirurgie élective des sinus maxillaires et ethmoïdaux, les concentrations médianes respectives de cefdinir dans le tissu sinusal 4 heures après l'administration de doses uniques de 300 et 600 mg étaient
Tissu pulmonaire
Chez les patients adultes subissant une bronchoscopie diagnostique, les concentrations médianes respectives de cefdinir dans la muqueuse bronchique 4 heures après l'administration de doses uniques de 300 et 600 mg étaient de 0,78 (
Liquide de l'oreille moyenne
Chez 14 patients pédiatriques atteints d'otite moyenne bactérienne aiguë, les concentrations médianes respectives de cefdinir dans le liquide de l'oreille moyenne 3 heures après l'administration de doses uniques de 7 et 14 mg/kg étaient de 0,21 (
FSC
Les données sur la pénétration du cefdinir dans le liquide céphalo-rachidien humain ne sont pas disponibles.
Métabolisme et excrétion
Le cefdinir n'est pas métabolisé de manière appréciable. L'activité est principalement due à la molécule mère. Le cefdinir est principalement éliminé par excrétion rénale avec une demi-vie d'élimination plasmatique moyenne (t½) de 1,7 (± 0,6) heures. Chez les sujets sains ayant une fonction rénale normale, la clairance rénale est de 2,0 (± 1,0) ml/min/kg et la clairance orale apparente est de 11,6 (± 6,0) et 15,5 (± 5,4) ml/min/kg après des doses de 300 et 600 -mg, respectivement. Le pourcentage moyen de dose récupérée inchangée dans l'urine après des doses de 300 et 600 mg est de 18,4 % (± 6,4) et 11,6 % (± 4,6), respectivement. La clairance du cefdinir est réduite chez les patients présentant une insuffisance rénale (voir Populations particulières - Patients insuffisants rénaux ).
Étant donné que l'excrétion rénale est la voie d'élimination prédominante, la posologie doit être ajustée chez les patients dont la fonction rénale est nettement altérée ou qui sont sous hémodialyse (voir DOSAGE ET ADMINISTRATION ).
Populations particulières
Patients insuffisants rénaux
La pharmacocinétique du cefdinir a été étudiée chez 21 sujets adultes présentant divers degrés de fonction rénale. Les diminutions du taux d'élimination du cefdinir, de la clairance orale apparente (CL/F) et de la clairance rénale étaient approximativement proportionnelles à la réduction de la clairance de la créatinine (CLcr). En conséquence, les concentrations plasmatiques de cefdinir étaient plus élevées et persistaient plus longtemps chez les sujets insuffisants rénaux que chez ceux sans insuffisance rénale. Chez les sujets avec une CLcr entre 30 et 60 mL/min, la Cmax et la t½ ont augmenté d'environ 2 fois et l'ASC d'environ 3 fois. Chez les sujets avec une CLcr DOSAGE ET ADMINISTRATION ).
Hémodialyse
La pharmacocinétique du cefdinir a été étudiée chez 8 sujets adultes sous hémodialyse. La dialyse (durée de 4 heures) a éliminé 63 % du cefdinir de l'organisme et réduit la t½ d'élimination apparente de 16 (± 3,5) à 3,2 (± 1,2) heures. Un ajustement posologique est recommandé dans cette population de patients (voir DOSAGE ET ADMINISTRATION ).
Maladie hépatique
Étant donné que le cefdinir est principalement éliminé par voie rénale et n'est pas métabolisé de manière appréciable, aucune étude n'a été menée chez des patients atteints d'insuffisance hépatique. On ne s'attend pas à ce qu'un ajustement posologique soit nécessaire dans cette population.
Patients gériatriques
L'effet de l'âge sur la pharmacocinétique du cefdinir après une dose unique de 300 mg a été évalué chez 32 sujets âgés de 19 à 91 ans. L'exposition systémique au cefdinir était considérablement augmentée chez les sujets âgés (N = 16), la Cmax de 44 % et l'ASC de 86 %. Cette augmentation était due à une diminution de la clairance du cefdinir. Le volume apparent de distribution a également été réduit, donc aucune modification appréciable de la t½ d'élimination apparente n'a été observée (personnes âgées : 2,2 ± 0,6 heures vs jeunes : 1,8 ± 0,4 heures). Étant donné qu'il a été démontré que la clairance du cefdinir est principalement liée à des modifications de la fonction rénale plutôt qu'à l'âge, les patients âgés ne nécessitent pas d'ajustement posologique à moins que leur fonction rénale ne soit nettement altérée (clairance de la créatinine Patients insuffisants rénaux, ci-dessus ).
Genre et race
Les résultats d'une méta-analyse de la pharmacocinétique clinique (N = 217) n'ont indiqué aucun impact significatif du sexe ou de la race sur la pharmacocinétique du cefdinir.
Microbiologie
Mécanisme d'action
Comme pour les autres céphalosporines, l'activité bactéricide du cefdinir résulte de l'inhibition de la synthèse de la paroi cellulaire. Le cefdinir est stable en présence de certaines enzymes β-lactamases, mais pas de toutes. En conséquence, de nombreux organismes résistants aux pénicillines et à certaines céphalosporines sont sensibles au cefdinir.
Mécanisme de résistance
La résistance au cefdinir est principalement due à l'hydrolyse par certaines β-lactamases, à l'altération des protéines de liaison à la pénicilline (PBP) et à la diminution de la perméabilité. Le cefdinir est inactif contre la plupart des souches d'Enterobacter spp., Pseudomonas spp., Enterococcus spp., streptocoques résistants à la pénicilline et staphylocoques résistants à la méthicilline. Les souches de H. influenzae négatives aux β-lactamases et résistantes à l'ampicilline (BLNAR) sont généralement non sensibles au cefdinir.
Activité anti-microbienne
Cefdinir s'est avéré actif contre la plupart des souches des micro-organismes suivants, à la fois in vitro et dans les infections cliniques, comme décrit dans INDICATIONS ET USAGE .
Bactéries Gram-positives
Staphylococcus aureus (souches sensibles à la méthicilline uniquement) Streptococcus pneumoniae (souches sensibles à la pénicilline uniquement) Streptococcus pyogenes
Bactéries Gram-négatives
Haemophilus influenzae Haemophilus parainfluenzae Moraxella catarrhalis
Les données in vitro suivantes sont disponibles, mais leur signification clinique est inconnue.
Le cefdinir présente des concentrations minimales inhibitrices (CMI) in vitro de 1 mcg/mL ou moins contre (≥ 90 %) les souches des micro-organismes suivants ; cependant, l'innocuité et l'efficacité du cefdinir dans le traitement des infections cliniques dues à ces micro-organismes n'ont pas été établies dans des essais cliniques adéquats et bien contrôlés.
Bactéries Gram-positives
Staphylococcus epidermidis (souches sensibles à la méthicilline uniquement) Streptococcus agalactiae Streptocoques du groupe Viridans
Bactéries Gram-négatives
Citrobacter koseri Escherichia coli Klebsiella pneumoniae Proteus mirabilis
Méthodes de test de sensibilité
Lorsqu'ils sont disponibles, le laboratoire de microbiologie clinique doit fournir des rapports périodiques décrivant le profil de sensibilité régional/local des agents pathogènes nosocomiaux et communautaires potentiels. Ces rapports devraient aider le médecin à choisir un médicament antibactérien pour le traitement.
Techniques de dilution
Des méthodes quantitatives sont utilisées pour déterminer les concentrations minimales inhibitrices (CMI) des antimicrobiens. Ces CMI fournissent des estimations de la sensibilité des bactéries aux composés antimicrobiens. Les CMI doivent être déterminées à l'aide d'une méthode de test standardisée1 (bouillon et/ou gélose). Les valeurs de CMI doivent être interprétées selon les critères fournis dans le tableau 1.
Techniques de diffusion
Les méthodes quantitatives qui nécessitent la mesure des diamètres de zone fournissent également des estimations reproductibles de la sensibilité des bactéries aux composés antimicrobiens. La taille de la zone doit être déterminée à l'aide d'une méthode normalisée.2 La procédure utilise des disques de papier imprégnés de 5 mcg de cefdinir pour tester la sensibilité des bactéries. Les critères d'interprétation de la diffusion du disque sont fournis dans le tableau 1.
La sensibilité des staphylocoques au cefdinir peut être déduite en testant la pénicilline et la céfoxitine ou l'oxacilline. Les staphylocoques sensibles à l'oxacilline (céfoxitine) peuvent être considérés comme sensibles au cefdinir.3
Un rapport « sensible » indique que l'antimicrobien est susceptible d'inhiber la croissance de l'agent pathogène si le composé antimicrobien atteint les concentrations au site d'infection nécessaires pour inhiber la croissance de l'agent pathogène. Un rapport "Intermédiaire" indique que le résultat doit être considéré comme équivoque et, si le micro-organisme n'est pas entièrement sensible aux médicaments alternatifs cliniquement réalisables, le test doit être répété. Cette catégorie implique une applicabilité clinique possible dans des sites corporels où le médicament est physiologiquement concentré ou dans des situations où une dose élevée de médicament peut être utilisée. Cette catégorie fournit également une zone tampon qui empêche les petits facteurs techniques incontrôlés de provoquer des divergences majeures dans l'interprétation. Un rapport de « résistant » indique que l'antimicrobien n'est pas susceptible d'inhiber la croissance de l'agent pathogène si le composé antimicrobien atteint les concentrations habituellement réalisables au site d'infection ; un autre traitement doit être choisi.
Contrôle de qualité
Les procédures de test de sensibilité standardisées nécessitent l'utilisation de contrôles de laboratoire pour surveiller et garantir l'exactitude et la précision des fournitures et des réactifs utilisés dans le test, ainsi que les techniques de la personne effectuant le test.1,2,3 La poudre de cefdinir standard doit fournir la plage suivante des valeurs de CMI comme indiqué dans le tableau 2. Pour la technique de diffusion utilisant un disque de 5 mcg, les critères du tableau 2 doivent être atteints.
Etudes cliniques
Pneumonie bactérienne acquise en communauté
Dans une étude contrôlée à double insu menée aux États-Unis chez des adultes et des adolescents, le cefdinir BID a été comparé au céfaclor 500 mg TID. En utilisant des critères stricts d'évaluabilité et de réponse microbiologique/clinique 6 à 14 jours après le traitement, les taux de guérison clinique, les taux d'éradication microbiologique présumés et les résultats statistiques suivants ont été obtenus :
Étude américaine sur la pneumonie communautaire Cefdinir vs Cefaclor
Dans une deuxième étude contrôlée à l'insu des investigateurs chez des adultes et des adolescents menée principalement en Europe, le cefdinir BID a été comparé à l'amoxicilline/acide clavulanique 500/125 mg TID. En utilisant des critères stricts d'évaluabilité et de réponse clinique 6 à 14 jours après le traitement, les taux de guérison clinique, les taux d'éradication microbiologique présumés et les résultats statistiques suivants ont été obtenus :
Étude sur la pneumonie acquise dans la communauté européenne Cefdinir vs Amoxicilline/Clavulanate
Pharyngite/amygdalite streptococcique
Dans quatre études contrôlées menées aux États-Unis, le cefdinir a été comparé à 10 jours de pénicilline chez des patients adultes, adolescents et pédiatriques. Deux études (une chez des adultes et des adolescents, l'autre chez des patients pédiatriques) ont comparé 10 jours de cefdinir QD ou BID à la pénicilline 250 mg ou 10 mg/kg QID. En utilisant des critères stricts d'évaluabilité et de réponse microbiologique/clinique 5 à 10 jours après le traitement, les taux de guérison clinique, les taux d'éradication microbiologique et les résultats statistiques suivants ont été obtenus :
Études sur la pharyngite/amygdalite Cefdinir (10 jours) vs pénicilline (10 jours)
Deux études (une chez des adultes et des adolescents, l'autre chez des patients pédiatriques) ont comparé 5 jours de cefdinir BID à 10 jours de pénicilline 250 mg ou 10 mg/kg QID. En utilisant des critères stricts d'évaluabilité et de réponse microbiologique/clinique 4 à 10 jours après le traitement, les taux de guérison clinique, les taux d'éradication microbiologique et les résultats statistiques suivants ont été obtenus :
Études sur la pharyngite/amygdalite Cefdinir (5 jours) vs pénicilline (10 jours)
RÉFÉRENCES
1. Institut des normes cliniques et de laboratoire (CLSI). Méthodes de dilution des tests de sensibilité aux antimicrobiens pour les bactéries à croissance aérobie ; Norme approuvée – Dixième édition. Document CLSI M07-A10 [2015], Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvanie 19087, États-Unis.
2. Institut des normes cliniques et de laboratoire (CLSI). Normes de performance pour les tests de sensibilité à la diffusion de disques antimicrobiens ; Norme approuvée – Douzième édition. Document CLSI M02-A12 [2015], Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvanie 19087, États-Unis.
3. Institut des normes cliniques et de laboratoire (CLSI). Normes de performance pour les tests de sensibilité aux antimicrobiens ; Vingt-cinquième supplément d'information, document CLSI M100-S25 [2015], Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvanie 19087, États-Unis.
4. Cockcroft DW, Gault MH. Prédiction de la clairance de la créatinine à partir de la créatinine sérique. Néphron 1976;16:31-41.
5. Schwartz GJ, Haycock GB, Edelmann CM, Spitzer A. Une simple estimation du taux de filtration glomérulaire chez les enfants dérivée de la longueur du corps et de la créatinine plasmatique. Pédiatrie 1976;58:259-63.
6. Schwartz GJ, Feld LG, Langford DJ. Une estimation simple du taux de filtration glomérulaire chez les nourrissons nés à terme au cours de la première année de vie. J Pediatrics 1984;104:849-54.
INFORMATIONS PATIENTS
Les patients doivent être informés que les médicaments antibactériens, y compris OMNICEF 300 mg, ne doivent être utilisés que pour traiter les infections bactériennes. Ils ne traitent pas les infections virales (par exemple, le rhume). Lorsqu'OMNICEF 300 mg est prescrit pour traiter une infection bactérienne, les patients doivent être informés que bien qu'il soit courant de se sentir mieux au début du traitement, le médicament doit être pris exactement comme indiqué. Sauter des doses ou ne pas terminer le traitement complet peut (1) diminuer l'efficacité du traitement immédiat et (2) augmenter la probabilité que les bactéries développent une résistance et ne puissent pas être traitées par OMNICEF 300 mg ou d'autres médicaments antibactériens à l'avenir.
Les antiacides contenant du magnésium ou de l'aluminium interfèrent avec l'absorption du cefdinir. Si ce type d'antiacide est nécessaire pendant le traitement par OMNICEF 300 mg, OMNICEF doit être pris au moins 2 heures avant ou après l'antiacide.
Les suppléments de fer, y compris les multivitamines contenant du fer, interfèrent avec l'absorption du cefdinir. Si des suppléments de fer sont nécessaires pendant le traitement par OMNICEF 300 mg, OMNICEF doit être pris au moins 2 heures avant ou après le supplément.
Les préparations pour nourrissons enrichies de fer n'interfèrent pas significativement avec l'absorption du cefdinir. Par conséquent, OMNICEF 300 mg pour suspension buvable peut être administré avec une préparation pour nourrissons enrichie en fer.
Les patients diabétiques et les soignants doivent savoir que la suspension buvable contient 2,86 g de saccharose par cuillère à café.
La diarrhée est un problème courant causé par les antibiotiques qui se termine généralement lorsque l'antibiotique est arrêté. Parfois, après le début du traitement aux antibiotiques, les patients peuvent développer des selles liquides et sanglantes (avec ou sans crampes d'estomac et fièvre) même deux mois ou plus après avoir pris la dernière dose de l'antibiotique. Si cela se produit, les patients doivent contacter leur médecin dès que possible.