Vantin 200mg, 100mg Cefpodoxime Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce que Vantin 100 mg et comment est-il utilisé ?
Vantin 100 mg est un médicament sur ordonnance utilisé pour traiter les symptômes de diverses infections bactériennes telles que la bronchite, la pneumonie, la sinusite, la pharyngite, l'amygdalite, les infections des voies urinaires, les infections cutanées et la gonorrhée. Vantin 200 mg peut être utilisé seul ou avec d'autres médicaments.
Vantin appartient à une classe de médicaments appelés caphalosporines, 3e génération.
On ne sait pas si Vantin est sûr et efficace chez les enfants de moins de 2 mois.
Quels sont les effets secondaires possibles de Vantin 200 mg ?
Vantin peut provoquer des effets secondaires graves, notamment :
- urticaire,
- difficulté à respirer,
- gonflement du visage, des lèvres, de la langue ou de la gorge,
- fièvre,
- mal de gorge,
- brûlant dans tes yeux,
- douleurs cutanées,
- éruption cutanée rouge ou violette qui se propage et provoque des cloques et une desquamation,
- fortes douleurs à l'estomac,
- diarrhée aqueuse ou sanglante,
- glandes enflées,
- douleur articulaire,
- malaise général,
- des battements de cœur battants,
- flottant dans ta poitrine,
- essoufflement, et
- saisie
Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires les plus courants de Vantin incluent :
- démangeaisons ou pertes vaginales,
- érythème fessier chez un nourrisson utilisant ce médicament,
- nausée,
- vomissement,
- diarrhée,
- douleurs à l'estomac et
- mal de tête
Dites au médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles de Vantin. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
Pour réduire le développement de bactéries résistantes aux médicaments et maintenir l'efficacité de VANTIN 100 mg et d'autres médicaments antibactériens, VANTIN 100 mg doit être utilisé uniquement pour traiter ou prévenir les infections dont l'origine bactérienne est avérée ou fortement suspectée.
LA DESCRIPTION
Le cefpodoxime proxétil est un antibiotique semi-synthétique à spectre étendu administré par voie orale de la classe des céphalosporines. Le nom chimique est (RS)-1(isopropoxycarbonyloxy)éthyl (+)-(6R,7R)-7-[2-(2-amino-4-thiazolyl)-2-{(Z)methoxyimino}acetamido]-3 -méthoxyméthyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ène-2-carboxylate. Sa formule empirique est C21H27N5O9S2 et sa formule structurale est représentée ci-dessous :
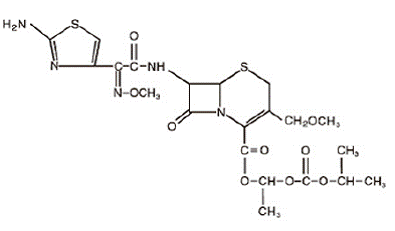
Le poids moléculaire du cefpodoxime proxétil est de 557,6.
Le cefpodoxime proxétil est un promédicament ; son métabolite actif est le cefpodoxime. Toutes les doses de cefpodoxime proxétil dans cette notice sont exprimées en termes de fraction active cefpodoxime. Le médicament est fourni à la fois sous forme de comprimés pelliculés et de granulés aromatisés pour suspension buvable.
Les comprimés VANTIN contiennent du cefpodoxime proxétil équivalent à 100 mg ou 200 mg d'activité de cefpodoxime et les ingrédients inactifs suivants : carboxyméthylcellulose calcique, cire de carnauba, FD&C jaune n° 6, hydroxypropylcellulose, hypromellose, lactose hydraté, stéarate de magnésium, propylène glycol, laurylsulfate de sodium et le dioxyde de titane. De plus, les comprimés pelliculés à 100 mg contiennent du D&C Jaune n° 10 et les comprimés pelliculés à 200 mg contiennent du FD&C Rouge n° 40.
Chaque 5 mL de suspension orale VANTIN contient du cefpodoxime proxétil équivalent à 50 mg ou 100 mg d'activité de cefpodoxime après constitution et les ingrédients inactifs suivants : arômes artificiels, hydroxy anisole butylé (BHA), carboxyméthylcellulose sodique, cellulose microcristalline, carraghénane, acide citrique, colloïdal dioxyde de silicium, croscarmellose sodique, hydroxypropylcellulose, lactose, maltodextrine, arômes naturels, alginate de propylène glycol, citrate de sodium, benzoate de sodium, amidon, saccharose et huile végétale.
LES INDICATIONS
Le cefpodoxime proxétil est indiqué pour le traitement des patients atteints d'infections légères à modérées causées par des souches sensibles des micro-organismes désignés dans les conditions énumérées ci-dessous.
Les posologies recommandées, les durées de traitement et les populations de patients applicables varient selon ces infections. Veuillez consulter POSOLOGIE ET ADMINISTRATION pour des recommandations spécifiques. Otite moyenne aiguë causée par Streptococcus pneumoniae (à l'exclusion des souches résistantes à la pénicilline), Streptococcus pyogenes, Haemophilus influenzae (y compris les souches productrices de bêta-lactamases) ou Moraxella (Branhamella) catarrhalis (y compris les souches productrices de bêta-lactamases).
Pharyngite et/ou amygdalite causée par Streptococcus pyogenes.
REMARQUE: Seule la pénicilline administrée par voie intramusculaire s'est révélée efficace dans la prophylaxie du rhumatisme articulaire aigu. Le cefpodoxime proxétil est généralement efficace dans l'éradication des streptocoques de l'oropharynx. Cependant, les données établissant l'efficacité du cefpodoxime proxetil pour la prophylaxie du rhumatisme articulaire aigu ultérieur ne sont pas disponibles.
Pneumonie communautaire causée par S. pneumoniae ou H. Influenzae (y compris les souches produisant des bêta-lactamases).
Exacerbation bactérienne aiguë de la bronchite chronique causée par S. pneumoniae, H. influenzae (souches non productrices de bétalactamase uniquement) ou M. catarrhalis. Les données sont actuellement insuffisantes pour établir l'efficacité chez les patients présentant des exacerbations bactériennes aiguës de bronchite chronique causées par des souches productrices de bêta-lactamase de H. influenzae.
Gonorrhée urétrale et cervicale aiguë non compliquée causée par Neisseria gonorrhoeae (y compris les souches productrices de pénicillinase).
Infections ano-rectales aiguës non compliquées chez la femme due à Neisseria gonorrhoeae (y compris les souches productrices de pénicillinase).
REMARQUE: L'efficacité de la cefpodoxime dans le traitement des patients de sexe masculin atteints d'infections rectales causées par N. gonorrhoeae n'a pas été établie. Les données ne supportent pas l'utilisation du cefpodoxime proxétil dans le traitement des infections pharyngées dues à N. gonorrhoeae chez l'homme ou la femme.
Infections non compliquées de la peau et des tissus cutanés causée par Staphylococcus aureus (y compris les souches productrices de pénicillinase) ou Streptococcus pyogenes. Les abcès doivent être drainés chirurgicalement selon les indications cliniques.
REMARQUE: Dans les essais cliniques, la réussite du traitement des infections non compliquées de la peau et des structures cutanées était liée à la dose. La dose thérapeutique efficace pour les infections cutanées était supérieure à celles utilisées dans les autres indications recommandées. (Voir DOSAGE ET ADMINISTRATION .)
Sinusite maxillaire aiguë causée par Haemophilus influenzae (y compris les souches productrices de bêta-lactamases), Streptococcus pneumoniae et Moraxella catarrhalis.
Infections urinaires non compliquées (cystite) causée par Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis ou Staphylococcus saprophyticus.
REMARQUE: Lors de l'examen de l'utilisation du cefpodoxime proxétil dans le traitement de la cystite, les taux d'éradication bactérienne inférieurs du cefpodoxime proxétil doivent être mis en balance avec les taux d'éradication accrus et les différents profils d'innocuité de certaines autres classes d'agents approuvés. (Voir Etudes cliniques section.)
Des échantillons appropriés pour un examen bactériologique doivent être obtenus afin d'isoler et d'identifier les organismes responsables et de déterminer leur sensibilité au cefpodoxime. Une thérapie peut être instaurée en attendant les résultats de ces études. Une fois ces résultats disponibles, l'antibiothérapie doit être adaptée en conséquence.
Pour réduire le développement de bactéries résistantes aux médicaments et maintenir l'efficacité de VANTIN 100 mg et d'autres médicaments antibactériens, VANTIN 200 mg doit être utilisé uniquement pour traiter ou prévenir les infections dont il est prouvé ou fortement suspecté qu'elles sont causées par des bactéries sensibles. Lorsque des informations sur la culture et la sensibilité sont disponibles, elles doivent être prises en compte lors de la sélection ou de la modification du traitement antibactérien. En l'absence de telles données, l'épidémiologie locale et les schémas de sensibilité peuvent contribuer à la sélection empirique du traitement.
DOSAGE ET ADMINISTRATION
(Voir INDICATIONS ET USAGE pour les agents pathogènes indiqués.)
Comprimés pelliculés
Les comprimés VANTIN doivent être administrés par voie orale avec de la nourriture pour améliorer l'absorption. (Voir PHARMACOLOGIE CLINIQUE .)
Les posologies recommandées, les durées de traitement et la population de patients applicable sont décrites dans le tableau suivant :
Adultes et adolescents (12 ans et plus)
Granulés pour suspension orale
La suspension orale VANTIN peut être administrée sans égard aux aliments. Les posologies recommandées, les durées de traitement et les populations de patients applicables sont décrites dans le tableau suivant :
Adultes et adolescents (12 ans et plus)
Nourrissons et patients pédiatriques (âgés de 2 mois à 12 ans)
Patients atteints de dysfonction rénale
Pour les patients présentant une insuffisance rénale sévère (clairance de la créatinine
Lorsque seul le taux de créatinine sérique est disponible, la formule suivante (basée sur le sexe, le poids et l'âge du patient) peut être utilisée pour estimer la clairance de la créatinine (mL/min). Pour que cette estimation soit valide, le taux de créatinine sérique doit représenter un état stable de la fonction rénale.
Hommes (mL/min) : Poids (kg) x (140 - âge)/ 72 x créatinine sérique (mg/100 mL)
Femelles(mL/min) : 0,85 x valeur supérieure
Patients atteints de cirrhose
La pharmacocinétique de la cefpodoxime chez les patients cirrhotiques (avec ou sans ascite) est similaire à celle des sujets sains. Aucun ajustement posologique n'est nécessaire dans cette population.
Préparation de la suspension
Directives constitutionnelles pour la suspension orale
Après mélange, la suspension doit être conservée au réfrigérateur, entre 2° et 8°C (36° et 46°F). Bien agiter avant utilisation. Conserver le récipient bien fermé. Le mélange peut être utilisé pendant 14 jours. Jeter la portion inutilisée après 14 jours.
COMMENT FOURNIE
VANTIN Comprimés sont disponibles dans les dosages (équivalent cefpodoxime), couleurs et tailles suivants :
100 mg (orange clair, elliptique, gravé avec U3617)
Bouteilles de 20 CDN 0009-3617-01 Flacons de 100 CDN 0009-3617-02 Doses unitaires de 100 CDN 0009-3617-03
200 mg (rouge corail, elliptique, gravé avec U3618)
Bouteilles de 20 CDN 0009-3618-01 Flacons de 100 CDN 0009-3618-02
200 mg (rouge corail, elliptique, gravé avec U3618)
Doses unitaires de 100 CDN 0009-3618-03
Conservez les comprimés à température ambiante contrôlée entre 20 et 25 °C (68 et 77 °F) [voir USP ].
Bien remettre le capuchon après chaque ouverture. Protégez les packs de doses unitaires de l'humidité excessive.
VANTIN 200mg Suspension buvable fournit l'équivalent de 50 mg ou 100 mg de cefpodoxime par suspension de 5 ml (lorsqu'il est constitué selon les instructions) et est disponible en saveur de crème au citron dans les tailles suivantes :
50 mg/5 ml
suspension de 100 ml CDN 0009-3531-01 suspension de 75 ml CDN 0009-3531-02 suspension de 50 ml CDN 0009-3531-03
100 mg/5 ml
suspension de 100 ml CDN 0009-3615-01 Suspension de 75 ml CDN 0009-3615-02 suspension de 50 ml CDN 0009-3615-03
Conservez les granulés non suspendus à température ambiante contrôlée entre 20 et 25 °C (68 et 77 °F) [voir USP ].
Les instructions de mélange sont incluses sur l'étiquette. Après mélange, la suspension doit être conservée au réfrigérateur, entre 2° et 8°C (36° et 46°F). Bien agiter avant utilisation. Conserver le récipient bien fermé. Le mélange peut être utilisé pendant 14 jours. Jeter la portion inutilisée après 14 jours.
Distribué par : Pharmacia & Upjohn Company, Division of Pfizer Inc., NY, NY 10017. Révisé : août 2016
EFFETS SECONDAIRES
Essais cliniques
Comprimés pelliculés (doses multiples)
Dans les essais cliniques utilisant doses multiples de cefpodoxime proxétil comprimés pelliculés, 4696 patients ont été traités avec les posologies recommandées de cefpodoxime (100 à 400 mg Q 12 heures). Il n'y a eu aucun décès ni incapacité permanente liés à la toxicité des médicaments. Cent vingt-neuf (2,7 %) patients ont arrêté le traitement en raison d'effets indésirables que l'on croyait possiblement ou probablement liés à la toxicité du médicament. Quatre-vingt-treize (52 %) des 178 patients qui ont interrompu le traitement (que l'on pense qu'il soit lié ou non au traitement médicamenteux) l'ont fait en raison de troubles gastro-intestinaux, de nausées, de vomissements ou de diarrhée. Le pourcentage de patients traités par cefpodoxime proxétil qui ont arrêté le médicament à l'étude en raison d'événements indésirables était significativement plus élevé à une dose de 800 mg par jour qu'à une dose de 400 mg par jour ou à une dose de 200 mg par jour. Les événements indésirables considérés comme possiblement ou probablement liés à la cefpodoxime dans les essais cliniques à doses multiples (N = 4 696 patients traités par la cefpodoxime) étaient :
Incidence supérieure à 1 %
Diarrhée 7,0 %
La diarrhée ou les selles molles étaient liées à la dose : passant de 10,4 % des patients recevant 800 mg par jour à 5,7 % pour ceux recevant 200 mg par jour. Parmi les patients souffrant de diarrhée, 10 % présentaient un organisme ou une toxine C. difficile dans les selles. (Voir AVERTISSEMENTS .)
Nausées 3,3 % Infections fongiques vaginales 1,0 % Infections vulvo-vaginales 1,3 % Douleurs abdominales 1,2 % Céphalées 1,0 %
Incidence inférieure à 1 %
Par système corporel par ordre décroissant
Etudes cliniques
Événements indésirables considérés comme possiblement ou probablement liés au cefpodoxime proxétil survenus chez moins de 1 % des patients (N = 4 696)
Corps - infections fongiques, distension abdominale, malaise, fatigue, asthénie, fièvre, douleur thoracique, dorsalgie, frissons, douleur généralisée, tests microbiologiques anormaux, candidose, abcès, réaction allergique, œdème facial, infections bactériennes, infections parasitaires, œdème localisé, douleur localisée .
Cardiovasculaire - insuffisance cardiaque congestive, migraine, palpitations, vasodilatation, hématome, hypertension, hypotension.
Digestif - vomissements, dyspepsie, bouche sèche, flatulences, diminution de l'appétit, constipation, candidose buccale, anorexie, éructation, gastrite, ulcères buccaux, troubles gastro-intestinaux, troubles rectaux, troubles de la langue, troubles dentaires, soif accrue, lésions buccales, ténesme, gorge sèche, mal de dents .
Hémique et Lymphatique - anémie.
Métabolique et Nutritionnel - déshydratation, goutte, œdème périphérique, prise de poids.
Musculo-squelettique - myalgie.
Nerveux - étourdissements, insomnie, somnolence, anxiété, tremblements, nervosité, infarctus cérébral, modification des rêves, troubles de la concentration, confusion, cauchemars, paresthésie, vertiges.
Respiratoire - asthme, toux, épistaxis, rhinite, respiration sifflante, bronchite, dyspnée, épanchement pleural, pneumonie, sinusite.
Peau - urticaire, éruption cutanée, prurit au site de non-application, diaphorèse, éruption maculopapulaire, dermatite fongique, desquamation, peau sèche au site de non-application, chute des cheveux, éruption vésiculobulleuse, coup de soleil.
Sens spéciaux - altérations du goût, irritation des yeux, perte du goût, acouphènes.
Urogénital - hématurie, infections des voies urinaires, métrorragies, dysurie, pollakiurie, nycturie, infection pénienne, protéinurie, douleurs vaginales.
Granulés pour suspension orale (doses multiples)
Dans des essais cliniques utilisant des doses multiples de granulés de cefpodoxime proxétil pour suspension buvable, 2128 patients pédiatriques (dont 93 % étaient âgés de moins de 12 ans) ont été traités avec les doses recommandées de cefpodoxime (10 mg/kg/jour toutes les 24 heures ou fractionné Q 12 heures à une dose adulte équivalente maximale). Il n'y a eu aucun décès ou incapacité permanente chez aucun des patients de ces études. Vingt-quatre patients (1,1 %) ont arrêté le traitement en raison d'effets indésirables que l'on croyait possiblement ou probablement liés au médicament à l'étude. Ces arrêts concernaient principalement des troubles gastro-intestinaux, généralement des diarrhées, des vomissements ou des éruptions cutanées.
Les événements indésirables jugés possiblement ou probablement liés, ou de relation inconnue, au cefpodoxime proxétil pour suspension buvable dans les essais cliniques à doses multiples (N = 2128 patients traités par cefpodoxime) étaient :
Incidence supérieure à 1 %
Diarrhée 6,0 % L'incidence de la diarrhée chez les nourrissons et les tout-petits (âgés de 1 mois à 2 ans) était de 12,8 %. Érythème fessier/éruption cutanée fongique 2,0 % (y compris la candidose) L'incidence de l'érythème fessier chez les nourrissons et les tout-petits était de 8,5 %. Autres éruptions cutanées 1,8 % Vomissements 2,3 %
Incidence inférieure à 1 %
Corps: Douleurs abdominales localisées, crampes abdominales, céphalées, monilia, douleurs abdominales généralisées, asthénie, fièvre, infection fongique.
Digestif: Nausées, monilia, anorexie, bouche sèche, stomatite, colite pseudomembraneuse.
Hémique & Lymphatique : Thrombocytémie, test de Coombs direct positif, éosinophilie, hyperleucocytose, leucopénie, allongement du temps de thromboplastine partielle, purpura thrombocytopénique.
Métabolique & Nutritionnel : Augmentation du SGPT.
Musculo-squelettique : Myalgie.
Nerveux: Hallucinations, hyperkinésie, nervosité, somnolence.
Respiratoire: Épistaxis, rhinite.

Peau: Candidose cutanée, urticaire, dermatite fongique, acné, dermatite exfoliative, éruption maculopapuleuse.
Sens spéciaux : Perversion du goût.
Comprimés pelliculés (dose unique)
Dans les essais cliniques utilisant un une seule dose de cefpodoxime proxétil comprimés pelliculés, 509 patients ont été traités avec la posologie recommandée de cefpodoxime (200 mg). Il n'y a eu aucun décès ni incapacité permanente liés à la toxicité des médicaments dans ces études.
Les événements indésirables considérés comme possiblement ou probablement liés au cefpodoxime dans les essais cliniques à dose unique menés aux États-Unis étaient :
Incidence supérieure à 1 %
Nausées 1,4 % Diarrhée 1,2 %
Incidence inférieure à 1 %
Système nerveux central : étourdissements, maux de tête, syncope. Dermatologique : éruption cutanée. Génital : Vaginite. Gastro-intestinal : douleurs abdominales. Psychiatrique : Anxiété.
Changements de laboratoire
Les changements de laboratoire significatifs qui ont été rapportés chez les patients adultes et pédiatriques dans les essais cliniques du cefpodoxime proxétil, sans égard à la relation médicamenteuse, étaient :
Hépatique: Augmentations transitoires de l'AST (SGOT), de l'ALT (SGPT), de la GGT, de la phosphatase alcaline, de la bilirubine et de la LDH.
Hématologique : Éosinophilie, leucocytose, lymphocytose, granulocytose, basophilie, monocytose, thrombocytose, diminution de l'hémoglobine, diminution de l'hématocrite, leucopénie, neutropénie, lymphocytopénie, thrombocytopénie, thrombocytémie, test de Coombs positif et prolongation du TP et du PTT.
Chimie sérique : Hyperglycémie, hypoglycémie, hypoalbuminémie, hypoprotéinémie, hyperkaliémie et hyponatrémie.
Rénal: Augmentation du BUN et de la créatinine.
La plupart de ces anomalies étaient transitoires et non cliniquement significatives.
Expérience post-commercialisation
Les effets indésirables graves suivants ont été signalés : réactions allergiques, y compris syndrome de Stevens-Johnson, nécrolyse épidermique toxique, érythème polymorphe et réactions de type maladie sérique, colite pseudomembraneuse, diarrhée sanglante avec douleur abdominale, rectocolite hémorragique, rectorragie avec hypotension, choc anaphylactique, lésions hépatiques, exposition in utero avec fausse couche, néphrite purpurique, infiltrat pulmonaire avec éosinophilie et dermatite des paupières.
Un décès a été attribué à une colite pseudomembraneuse et à une coagulation intravasculaire disséminée.
Étiquetage de la classe des céphalosporines
Outre les effets indésirables énumérés ci-dessus qui ont été observés chez des patients traités par le cefpodoxime proxétil, les effets indésirables suivants et les tests de laboratoire modifiés ont été rapportés pour les antibiotiques de la classe des céphalosporines :
Effets indésirables et tests de laboratoire anormaux : Dysfonctionnement rénal, néphropathie toxique, dysfonctionnement hépatique, y compris cholestase, anémie aplasique, anémie hémolytique, réaction de type maladie sérique, hémorragie, agranulocytose et pancytopénie.
Plusieurs céphalosporines ont été impliquées dans le déclenchement des crises, en particulier chez les insuffisants rénaux lorsque la posologie n'a pas été réduite. (Voir DOSAGE ET ADMINISTRATION et SURDOSAGE .) Si des convulsions associées au traitement médicamenteux se produisent, le médicament doit être interrompu. Un traitement anticonvulsivant peut être administré si cliniquement indiqué.
INTERACTIONS MÉDICAMENTEUSES
Antiacides
L'administration concomitante de fortes doses d'antiacides (bicarbonate de sodium et hydroxyde d'aluminium) ou d'anti-H2 réduit les concentrations plasmatiques maximales de 24 % à 42 % et le degré d'absorption de 27 % à 32 %, respectivement. Le taux d'absorption n'est pas modifié par ces médicaments concomitants. Les anticholinergiques oraux (p. ex., propanthéline) retardent les concentrations plasmatiques maximales (augmentation de 47 % du Tmax), mais n'affectent pas le degré d'absorption (ASC).
Probénécide
Comme avec d'autres antibiotiques bêta-lactamines, l'excrétion rénale du cefpodoxime a été inhibée par le probénécide et a entraîné une augmentation d'environ 31 % de l'ASC et une augmentation de 20 % des concentrations plasmatiques maximales de cefpodoxime.
Médicaments néphrotoxiques
Bien qu'aucune néphrotoxicité n'ait été notée lorsque le cefpodoxime proxétil était administré seul, une surveillance étroite de la fonction rénale est recommandée lorsque le cefpodoxime proxétil est administré en même temps que des composés au potentiel néphrotoxique connu.
Interactions médicament/test de laboratoire
Les céphalosporines, y compris le cefpodoxime proxétil, sont connues pour induire occasionnellement un test de Coombs direct positif.
AVERTISSEMENTS
AVANT QUE LA THÉRAPIE AVEC LE CEFPODOXIME PROXETIL SOIT INSTITUÉE, UNE ENQUÊTE PRUDENTE DEVRAIT ÊTRE FAITE POUR DÉTERMINER SI LE PATIENT A EU DES RÉACTIONS D'HYPERSENSIBILITÉ PRÉCÉDENTES AU CEFPODOXIME, À D'AUTRES CÉPHALOSPORINES, À DES PÉNICILLINES OU À D'AUTRES MÉDICAMENTS. SI LE CEFPODOXIME DOIT ÊTRE ADMINISTRÉ À DES PATIENTS SENSIBLES À LA PÉNICILLINE, LA PRUDENCE DOIT ÊTRE EXERCÉE CAR L'HYPERSENSIBILITÉ CROISÉE PARMI LES ANTIBIOTIQUES BÊTA-LACTAM A ÉTÉ CLAIREMENT DOCUMENTÉE ET PEUT SE PRODUIRE CHEZ JUSQU'À 10 % DES PATIENTS AYANT DES ANTÉCÉDENTS D'ALLERGIES À LA PÉNICILLINE. SI UNE RÉACTION ALLERGIQUE AU CEFPODOXIME PROXETIL SE PRODUIT, ARRÊTEZ LE MÉDICAMENT. LES RÉACTIONS D'HYPERSENSIBILITÉ AIGUË GRAVES PEUVENT NÉCESSAIRE UN TRAITEMENT AVEC DE L'ÉPINÉPHRINE ET D'AUTRES MESURES D'URGENCE, Y COMPRIS L'OXYGÈNE, LES LIQUIDES INTRAVEINEUSES, L'ANTHISTAMINE INTRAVEINEUSE ET LA GESTION DES VOIES RESPIRATOIRES, SELON L'INDICATION CLINIQUE.
Des diarrhées associées à Clostridium difficile (DACD) ont été signalées avec l'utilisation de presque tous les agents antibactériens, y compris VANTIN, et leur gravité peut varier d'une diarrhée légère à une colite mortelle. Le traitement avec des agents antibactériens altère la flore normale du côlon, entraînant une prolifération de C. difficile.
C. difficile produit les toxines A et B qui contribuent au développement de CDAD. Les souches de C. difficile productrices d'hypertoxines entraînent une morbidité et une mortalité accrues, car ces infections peuvent être réfractaires au traitement antimicrobien et nécessiter une colectomie. La DACD doit être envisagée chez tous les patients qui présentent une diarrhée suite à l'utilisation d'antibiotiques. Des antécédents médicaux minutieux sont nécessaires car il a été rapporté que la DACD survient plus de deux mois après l'administration d'agents antibactériens.
Si la DACD est suspectée ou confirmée, l'utilisation continue d'antibiotiques non dirigés contre C. difficile peut devoir être interrompue. Une gestion appropriée des liquides et des électrolytes, une supplémentation en protéines, un traitement antibiotique de C. difficile et une évaluation chirurgicale doivent être institués selon les indications cliniques.
Un effort concerté pour surveiller le C. difficile chez les patients atteints de diarrhée traités par cefpodoxime a été entrepris en raison d'une incidence accrue de diarrhée associée au C. difficile dans les premiers essais chez des sujets normaux. Des organismes ou une toxine C. difficile ont été signalés chez 10 % des patients adultes atteints de diarrhée traités par cefpodoxime; cependant, aucun diagnostic spécifique de colite pseudomembraneuse n'a été posé chez ces patients.
Dans l'expérience post-commercialisation en dehors des États-Unis, des cas de colite pseudomembraneuse associée à l'utilisation de cefpodoxime proxétil ont été signalés.
PRÉCAUTIONS
Général
Chez les patients présentant une réduction transitoire ou persistante du débit urinaire due à une insuffisance rénale, la dose quotidienne totale de cefpodoxime proxétil doit être réduite car des concentrations sériques élevées et prolongées d'antibiotiques peuvent survenir chez ces personnes après les doses habituelles. La cefpodoxime, comme les autres céphalosporines, doit être administrée avec prudence aux patients recevant un traitement concomitant par des diurétiques puissants. (Voir DOSAGE ET ADMINISTRATION .)
Comme avec d'autres antibiotiques, l'utilisation prolongée de cefpodoxime proxétil peut entraîner une prolifération d'organismes non sensibles. Une évaluation répétée de l'état du patient est essentielle. Si une surinfection survient pendant le traitement, des mesures appropriées doivent être prises.
La prescription de VANTIN en l'absence d'infection bactérienne avérée ou fortement suspectée ou d'indication prophylactique est peu susceptible d'apporter un bénéfice au patient et augmente le risque de développement de bactéries résistantes aux médicaments.
Carcinogenèse, mutagenèse, altération de la fertilité
Aucune étude de carcinogenèse animale à long terme sur le cefpodoxime proxétil n'a été réalisée. Les études de mutagenèse de la cefpodoxime, y compris le test d'Ames avec et sans activation métabolique, le test d'aberration chromosomique, le test de synthèse d'ADN non programmée, la recombinaison mitotique et la conversion génique, le test de mutation génique directe et le test du micronoyau in vivo, étaient toutes négatives. Aucun effet indésirable sur la fertilité ou la reproduction n'a été noté lorsque 100 mg/kg/jour ou moins (2 fois la dose humaine basée sur mg/m²) ont été administrés par voie orale à des rats.
Grossesse
Effets tératogènes
Le cefpodoxime proxétil n'a été ni tératogène ni embryocide lorsqu'il a été administré à des rats au cours de l'organogenèse à des doses allant jusqu'à 100 mg/kg/jour (2 fois la dose humaine en mg/m²) ou à des lapins à des doses allant jusqu'à 30 mg/kg/jour (1 -2 fois la dose humaine basée sur mg/m²).
Il n'existe cependant aucune étude adéquate et bien contrôlée sur l'utilisation du cefpodoxime proxétil chez la femme enceinte. Étant donné que les études sur la reproduction chez l'animal ne sont pas toujours prédictives de la réponse humaine, ce médicament ne doit être utilisé pendant la grossesse qu'en cas de nécessité absolue.
Travail et accouchement
Le cefpodoxime proxétil n'a pas été étudié pour une utilisation pendant le travail et l'accouchement. Le traitement ne doit être administré que s'il est clairement nécessaire.
Mères allaitantes
La cefpodoxime est excrétée dans le lait maternel. Dans une étude portant sur 3 femmes allaitantes, les taux de cefpodoxime dans le lait maternel étaient de 0 %, 2 % et 6 % des taux sériques concomitants 4 heures après une dose orale de 200 mg de cefpodoxime proxétil. Six heures après l'administration, les taux étaient de 0 %, 9 % et 16 % des taux sériques concomitants. En raison du risque de réactions graves chez les nourrissons, une décision doit être prise soit d'interrompre l'allaitement soit d'arrêter le médicament, en tenant compte de l'importance du médicament pour la mère.
Utilisation pédiatrique
L'innocuité et l'efficacité chez les nourrissons de moins de 2 mois n'ont pas été établies.
Utilisation gériatrique
Sur les 3338 patients ayant participé aux études cliniques à doses multiples de cefpodoxime proxétil comprimés pelliculés, 521 (16 %) avaient 65 ans et plus, tandis que 214 (6 %) avaient 75 ans et plus. Aucune différence globale d'efficacité ou de sécurité n'a été observée entre les patients âgés et les patients plus jeunes. Chez des sujets gériatriques sains ayant une fonction rénale normale, la demi-vie plasmatique de la cefpodoxime était en moyenne de 4,2 heures et la récupération urinaire était en moyenne de 21 % après l'administration d'une dose de 400 mg toutes les 12 heures pendant 15 jours. Les autres paramètres pharmacocinétiques sont restés inchangés par rapport à ceux observés chez les sujets sains plus jeunes.
Un ajustement posologique chez les patients âgés ayant une fonction rénale normale n'est pas nécessaire.
SURDOSAGE
Dans les études de toxicité aiguë chez les rongeurs, une dose orale unique de 5 g/kg n'a produit aucun effet indésirable.
En cas de réaction toxique grave due à un surdosage, l'hémodialyse ou la dialyse péritonéale peut aider à éliminer la cefpodoxime de l'organisme, en particulier si la fonction rénale est altérée.
Les symptômes toxiques consécutifs à une surdose d'antibiotiques bêta-lactamines peuvent inclure des nausées, des vomissements, une détresse épigastrique et de la diarrhée.
CONTRE-INDICATIONS
Le cefpodoxime proxétil est contre-indiqué chez les patients présentant une allergie connue au cefpodoxime ou aux antibiotiques du groupe des céphalosporines.
PHARMACOLOGIE CLINIQUE
Absorption et excrétion
Le cefpodoxime proxétil est un promédicament qui est absorbé par le tractus gastro-intestinal et désestérifié en son métabolite actif, le cefpodoxime. Après administration orale de 100 mg de cefpodoxime proxétil à des sujets à jeun, environ 50 % de la dose de cefpodoxime administrée a été absorbée par voie systémique. Dans l'intervalle posologique recommandé (100 à 400 mg), environ 29 à 33 % de la dose de cefpodoxime administrée ont été excrétés sous forme inchangée dans les urines en 12 heures. Le métabolisme de la cefpodoxime in vivo est minime.
Effets de la nourriture
Le degré d'absorption (ASC moyenne) et la concentration plasmatique maximale moyenne ont augmenté lorsque les comprimés pelliculés ont été administrés avec de la nourriture. Après une dose de comprimé de 200 mg prise avec de la nourriture, l'ASC était de 21 à 33 % supérieure à celle observée à jeun, et la concentration plasmatique maximale était en moyenne de 3,1 mcg/mL chez les sujets nourris contre 2,6 mcg/mL chez les sujets à jeun. Le temps nécessaire pour atteindre le pic de concentration n'était pas significativement différent entre les sujets nourris et à jeun.
Lorsqu'une dose de 200 mg de la suspension a été prise avec de la nourriture, le degré d'absorption (ASC moyenne) et la concentration plasmatique maximale moyenne chez les sujets nourris n'étaient pas significativement différents de ceux des sujets à jeun, mais le taux d'absorption était plus lent avec la nourriture (augmentation de 48 % en Tmax).
Pharmacocinétique des comprimés pelliculés de cefpodoxime proxétil
Dans la plage posologique recommandée (100 à 400 mg), la vitesse et l'étendue de l'absorption de la cefpodoxime étaient dose-dépendantes ; la Cmax et l'ASC normalisées en fonction de la dose ont diminué jusqu'à 32 % avec l'augmentation de la dose. Dans l'intervalle posologique recommandé, le Tmax était d'environ 2 à 3 heures et le T½ variait de 2,09 à 2,84 heures. La Cmax moyenne était de 1,4 mcg/mL pour la dose de 100 mg, de 2,3 mcg/mL pour la dose de 200 mg et de 3,9 mcg/mL pour la dose de 400 mg. Chez les patients ayant une fonction rénale normale, aucune accumulation ni modification significative des autres paramètres pharmacocinétiques n'a été notée après des doses orales multiples allant jusqu'à 400 mg toutes les 12 heures.
NIVEAUX PLASMATIQUES DE CEFPODOXIME (mcg/mL) CHEZ L'ADULTE À JEÛNE APRÈS L'ADMINISTRATION DE COMPRIMÉS PELLICULÉS (dose unique)
Pharmacocinétique de la suspension de cefpodoxime proxétil
Chez les sujets adultes, une dose de 100 mg de suspension buvable a produit un pic moyen de concentration de cefpodoxime d'environ 1,5 mcg/mL (intervalle : 1,1 à 2,1 mcg/mL), équivalent à celui rapporté après l'administration du comprimé de 100 mg. Le temps jusqu'au pic de concentration plasmatique et l'aire sous la courbe de concentration plasmatique en fonction du temps (ASC) pour la suspension buvable étaient également équivalents à ceux obtenus avec des comprimés pelliculés chez l'adulte après une dose orale de 100 mg.
La pharmacocinétique de la cefpodoxime a été étudiée chez 29 patients âgés de 1 à 17 ans. Chaque patient a reçu une dose orale unique de 5 mg/kg de suspension buvable de cefpodoxime. Des échantillons de plasma et d'urine ont été prélevés pendant 12 heures après l'administration. Les concentrations plasmatiques rapportées dans cette étude sont les suivantes :
NIVEAUX PLASMATIQUES DE CEFPODOXIME (mcg/mL) CHEZ LES PATIENTS À JEÛNEMENT (ÂGÉS DE 1 À 17 ANS) APRÈS L'ADMINISTRATION DE LA SUSPENSION
Distribution
La liaison aux protéines de la cefpodoxime varie de 22 à 33 % dans le sérum et de 21 à 29 % dans le plasma.
Peau Blister
Suite à l'administration de doses multiples toutes les 12 heures pendant 5 jours de 200 mg ou 400 mg de cefpodoxime proxétil, la concentration maximale moyenne de cefpodoxime dans le liquide des cloques cutanées était en moyenne de 1,6 et 2,8 mcg/mL, respectivement. Les niveaux de cefpodoxime dans le liquide vésical de la peau 12 heures après l'administration étaient en moyenne de 0,2 et 0,4 mcg/mL pour les régimes à doses multiples de 200 mg et 400 mg, respectivement.
Tissu d'amygdale
Après un seul comprimé pelliculé oral de 100 mg de cefpodoxime proxétil, la concentration maximale moyenne de cefpodoxime dans le tissu des amygdales était en moyenne de 0,24 mcg/g 4 heures après l'administration et de 0,09 mcg/g 7 heures après l'administration. L'équilibre a été atteint entre le plasma et le tissu des amygdales dans les 4 heures suivant l'administration. Aucune détection de cefpodoxime dans les tissus amygdaliens n'a été signalée 12 heures après l'administration. Ces résultats ont démontré que les concentrations de cefpodoxime dépassaient la CMI90 de S. pyogenes pendant au moins 7 heures après administration de 100 mg de cefpodoxime proxétil.
Tissu pulmonaire
Après un seul comprimé pelliculé oral de 200 mg de cefpodoxime proxétil, la concentration maximale moyenne de cefpodoxime dans le tissu pulmonaire était en moyenne de 0,63 mcg/g 3 heures après l'administration, de 0,52 mcg/g 6 heures après l'administration et de 0,19 mcg/g 12 heures après l'administration. Les résultats de cette étude ont indiqué que la cefpodoxime pénétrait dans les tissus pulmonaires et produisait des concentrations soutenues de médicament pendant au moins 12 heures après l'administration à des niveaux dépassant la CMI90 pour S. pneumoniae et H. influenzae.
FSC
Des données adéquates sur les taux de cefpodoxime dans le LCR ne sont pas disponibles.
Effets de la diminution de la fonction rénale
L'élimination de la cefpodoxime est réduite chez les patients présentant une insuffisance rénale modérée à sévère (clairance de la créatinine PRÉCAUTIONS et DOSAGE ET ADMINISTRATION .) Chez les sujets présentant une insuffisance rénale légère (clairance de la créatinine de 50 à 80 mL/min), la demi-vie plasmatique moyenne de la cefpodoxime était de 3,5 heures. Chez les sujets présentant une insuffisance rénale modérée (clairance de la créatinine de 30 à 49 ml/min) ou sévère (clairance de la créatinine de 5 à 29 ml/min), la demi-vie a augmenté à 5,9 et 9,8 heures, respectivement. Environ 23 % de la dose administrée a été éliminée de l'organisme au cours d'une procédure d'hémodialyse standard de 3 heures.
Effet de l'insuffisance hépatique (cirrhose)
L'absorption était quelque peu diminuée et l'élimination inchangée chez les patients atteints de cirrhose. La T½ moyenne de la cefpodoxime et la clairance rénale chez les patients cirrhotiques étaient similaires à celles obtenues dans les études sur des sujets sains. L'ascite n'a pas semblé affecter les valeurs chez les sujets cirrhotiques. Aucun ajustement posologique n'est recommandé dans cette population de patients.
Pharmacocinétique chez les sujets âgés
Les sujets âgés n'ont pas besoin d'ajustements posologiques sauf s'ils ont une fonction rénale diminuée. (Voir PRÉCAUTIONS .) Chez des sujets gériatriques sains, la demi-vie plasmatique de la cefpodoxime était en moyenne de 4,2 heures (contre 3,3 chez les sujets plus jeunes) et la récupération urinaire était en moyenne de 21 % après l'administration d'une dose de 400 mg toutes les 12 heures. Les autres paramètres pharmacocinétiques (Cmax, ASC et Tmax) sont restés inchangés par rapport à ceux observés chez les jeunes sujets sains.
Microbiologie
Mécanisme d'action
La cefpodoxime est un agent bactéricide qui agit par inhibition de la synthèse de la paroi cellulaire bactérienne. La cefpodoxime a une activité en présence de certaines bêta-lactamases, pénicillinases et céphalosporinases, de bactéries Gram-négatives et Gram-positives.
Mécanisme de résistance
La résistance à la cefpodoxime est principalement due à l'hydrolyse par la bêta-lactamase, à l'altération des protéines de liaison à la pénicilline (PBP) et à la diminution de la perméabilité.
La cefpodoxime s'est avérée active contre la plupart des isolats des bactéries suivantes, à la fois in vitro et dans les infections cliniques, comme décrit dans la section Indications et utilisation (1) :
Bactéries à Gram positif
Staphylococcus aureus (souches sensibles à la méthicilline, y compris celles produisant des pénicillinases) Staphylococcus saprophyticus Streptococcus pneumoniae (à l'exclusion des isolats résistants à la pénicilline) Streptococcus pyogenes
Bactéries Gram-négatives
Escherichia coli Klebsiella pneumoniae Proteus mirabilis Haemophilus influenzae (y compris les isolats producteurs de bêta-lactamase) Moraxella catarrhalis Neisseria gonorrhoeae (y compris les isolats producteurs de pénicillinase)
Les données in vitro suivantes sont disponibles, mais leur signification clinique est inconnue. Au moins 90 % des micro-organismes suivants présentent une concentration minimale inhibitrice (CMI) in vitro inférieure ou égale au point de rupture sensible pour la cefpodoxime. Cependant, l'efficacité de Cefpodoxime dans le traitement des infections cliniques dues à ces micro-organismes n'a pas été établie dans des essais cliniques adéquats et bien contrôlés.
Bactéries à Gram positif
Streptococcus agalactiae Streptococcus spp. (Groupes C, F, G)
Bactéries Gram-négatives
Citrobacter diversus Klebsiella oxytoca Proteus vulgaris Providencia rettgeri Haemophilus parainfluenzae
Bactéries anaérobies à Gram positif
Peptostreptococcus magnus
Méthodes de test de sensibilité
Lorsqu'ils sont disponibles, le laboratoire de microbiologie clinique doit fournir au médecin les résultats des tests de sensibilité in vitro pour les produits médicamenteux antimicrobiens utilisés dans les hôpitaux résidents sous forme de rapports périodiques décrivant le profil de sensibilité des agents pathogènes nosocomiaux et d'origine communautaire. Ces rapports devraient aider le médecin à choisir un médicament antibactérien pour le traitement.
Techniques de dilution
Des méthodes quantitatives sont utilisées pour déterminer les concentrations inhibitrices minimales antimicrobiennes (CMI). Ces CMI fournissent des estimations de la sensibilité des bactéries aux composés antimicrobiens. Les CMI doivent être déterminées à l'aide d'une méthode d'essai normalisée. Les valeurs de CMI doivent être interprétées selon les critères fournis dans le tableau 1.
Techniques de diffusion
Les méthodes quantitatives qui nécessitent la mesure des diamètres de zone fournissent également des estimations reproductibles de la sensibilité des bactéries aux composés antimicrobiens. La taille de la zone fournit une estimation de la sensibilité des bactéries aux composés antimicrobiens. La taille de la zone doit être déterminée à l'aide d'une méthode d'essai normalisée. Cette procédure utilise des disques de papier imprégnés de 10 mcg de Cefpodoxime pour tester la sensibilité des micro-organismes à la Cefpodoxime. Les critères d'interprétation de la diffusion du disque sont fournis dans le tableau 1.
Un rapport de Susceptible indique que l'antimicrobien est susceptible d'inhiber la croissance de l'agent pathogène si le composé antimicrobien atteint la concentration au site d'infection nécessaire pour inhiber la croissance de l'agent pathogène. Un rapport d'intermédiaire indique que le résultat doit être considéré comme équivoque, et si le micro-organisme n'est pas entièrement sensible aux médicaments alternatifs cliniquement réalisables, le test doit être répété. Cette catégorie implique une applicabilité clinique possible dans des sites corporels où le médicament est physiologiquement concentré ou dans des situations où une dose élevée de médicament peut être utilisée. Cette catégorie fournit également une zone tampon qui empêche les petits facteurs techniques incontrôlés de provoquer des divergences majeures dans l'interprétation. Un rapport de résistant indique que l'antimicrobien n'est pas susceptible d'inhiber la croissance de l'agent pathogène si le composé antimicrobien atteint les concentrations habituellement réalisables au site d'infection ; un autre traitement doit être choisi.
Contrôle de qualité
Les procédures de test de sensibilité standardisées nécessitent l'utilisation de contrôles de laboratoire pour surveiller et garantir l'exactitude et la précision des fournitures et des réactifs utilisés dans le test, ainsi que les techniques de la personne effectuant le test1,2,3. La poudre de cefpodoxime standard doit fournir la plage suivante de valeurs de CMI indiquées dans le tableau 2. Pour la technique de diffusion utilisant le disque de 10 mcg, les critères du tableau 2 doivent être atteints.
Essais cliniques
Cystite
Dans deux essais comparatifs randomisés en double aveugle 2:1 réalisés chez des adultes aux États-Unis, le cefpodoxime proxétil a été comparé à d'autres antibiotiques bêta-lactamines. Dans ces études, les taux d'éradication bactérienne suivants ont été obtenus 5 à 9 jours après le traitement :
Dans ces études, les taux de guérison clinique et les taux d'éradication bactérienne du cefpodoxime proxétil étaient comparables à ceux des agents de comparaison ; cependant, les taux de guérison clinique et les taux d'éradication bactériologique étaient inférieurs à ceux observés avec certaines autres classes d'agents approuvés pour la cystite.
Études sur l'otite moyenne aiguë
Dans des études contrôlées sur l'otite moyenne aiguë réalisées aux États-Unis, où des taux significatifs d'organismes producteurs de bêtalactamases ont été trouvés, le cefpodoxime proxétil a été comparé au céfixime. Dans ces études, en utilisant des critères d'évaluabilité très stricts et des critères de réponse microbiologique et clinique lors du suivi post-traitement de 4 à 21 jours, les résultats présomptifs suivants d'éradication bactérienne/succès clinique (guérison et amélioration) ont été obtenus.
RÉFÉRENCES
1. Institut des normes cliniques et de laboratoire (CLSI). Méthodes de dilution des tests de sensibilité aux antimicrobiens pour les bactéries à croissance aérobie ; Norme approuvée - Neuvième édition. Document CLSI M07-A9, Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvanie 19087, États-Unis, 2012.
2. Institut des normes cliniques et de laboratoire (CLSI). Normes de performance pour les tests de sensibilité aux antimicrobiens ; Vingt-troisième supplément d'information, document CLSI M100-S23. Document CLSI M100-S23, Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvanie 19087, États-Unis, 2013.
3. Institut des normes cliniques et de laboratoire (CLSI). Normes de performance pour les tests de sensibilité à la diffusion de disques antimicrobiens ; Norme approuvée - onzième édition du document CLSI M02-A11, Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvanie 19087, États-Unis, 2012.
INFORMATIONS PATIENTS
Les patients doivent être informés que les médicaments antibactériens, y compris VANTIN, ne doivent être utilisés que pour traiter les infections bactériennes. Ils ne traitent pas les infections virales (par exemple, le rhume). Lorsque VANTIN est prescrit pour traiter une infection bactérienne, les patients doivent être informés que même s'il est courant de se sentir mieux au début du traitement, le médicament doit être pris exactement comme indiqué. Sauter des doses ou ne pas terminer le traitement complet peut (1) diminuer l'efficacité du traitement immédiat et (2) augmenter la probabilité que les bactéries développent une résistance et ne puissent plus être traitées par VANTIN ou d'autres médicaments antibactériens à l'avenir.
La diarrhée est un problème courant causé par les antibiotiques qui se termine généralement lorsque l'antibiotique est arrêté. Parfois, après le début du traitement aux antibiotiques, les patients peuvent développer des selles liquides et sanglantes (avec ou sans crampes d'estomac et fièvre) même deux mois ou plus après avoir pris la dernière dose de l'antibiotique. Si cela se produit, les patients doivent contacter leur médecin dès que possible.