Arimidex 1mg Anastrozole Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce qu'Arimidex et comment est-il utilisé ?
Arimidex 1 mg est un médicament délivré sur ordonnance utilisé pour traiter les symptômes du cancer du sein chez les femmes ménopausées. Arimidex peut être utilisé seul ou avec d'autres médicaments.
Arimidex appartient à une classe de médicaments appelés antinéoplasiques, inhibiteurs de l'aromatase.
On ne sait pas si Arimidex est sûr et efficace chez les enfants.
Quels sont les effets secondaires possibles d'Arimidex ?
Arimidex peut provoquer des effets secondaires graves, notamment :
- essoufflement,
- fracture de l'os,
- glandes enflées,
- nausée,
- douleur dans le haut du ventre,
- démangeaison,
- fatigue,
- perte d'appétit,
- urine foncée,
- selles de couleur argile,
- jaunissement de la peau ou des yeux (jaunisse),
- engourdissement ou faiblesse soudaine (surtout d'un côté du corps),
- maux de tête intenses et soudains,
- troubles de l'élocution,
- problèmes de vision ou d'équilibre,
- fièvre,
- mal de gorge,
- gonflement du visage ou de la langue,
- brûlant dans tes yeux,
- douleurs cutanées et
- éruption cutanée rouge ou violette qui se propage (surtout au visage ou au haut du corps) avec cloques et desquamation
Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires les plus courants d'Arimidex comprennent :
- la faiblesse,
- les bouffées de chaleur,
- engourdissement ou sensation de picotement dans la peau,
- gonflement des chevilles ou des pieds,
- douleurs ou raideurs articulaires,
- problèmes avec vos doigts lors de la préhension,
- mal de gorge,
- mal de tête,
- mal au dos,
- douleur osseuse,
- la dépression,
- des changements d'humeur,
- troubles du sommeil (insomnie),
- hypertension artérielle,
- Maux de tête sévères,
- Vision floue,
- battant dans votre cou ou vos oreilles,
- nausée,
- des vomissements et
- légère éruption cutanée
Dites au médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles d'Arimidex. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
LA DESCRIPTION
Les comprimés ARIMIDEX (anastrozole) pour administration orale contiennent 1 mg d'anastrozole, un inhibiteur non stéroïdien de l'aromatase. Il est chimiquement décrit comme 1,3-benzènediacétonitrile, a, a, a', a'-tétraméthyl-5-(1H-1,2,4-triazol-1-ylméthyl). Sa formule moléculaire est C17H19N5 et sa formule développée est :
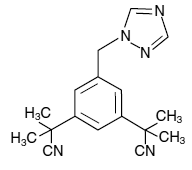
L'anastrozole est une poudre blanc cassé d'un poids moléculaire de 293,4. L'anastrozole a une solubilité aqueuse modérée (0,5 mg/mL à 25 °C); la solubilité est indépendante du pH dans la plage physiologique. L'anastrozole est librement soluble dans le méthanol, l'acétone, l'éthanol et le tétrahydrofurane, et très soluble dans l'acétonitrile.
Chaque comprimé contient comme ingrédients inactifs : lactose, stéarate de magnésium, hydroxypropylméthylcellulose, polyéthylèneglycol, povidone, glycolate d'amidon sodique et dioxyde de titane.
LES INDICATIONS
Traitement Adjuvant
ARIMIDEX est indiqué pour le traitement adjuvant des femmes ménopausées atteintes d'un cancer du sein précoce à récepteurs hormonaux positifs.
Traitement de première intention
ARIMIDEX 1 mg est indiqué dans le traitement de première intention des femmes ménopausées atteintes d'un cancer du sein localement avancé ou métastatique à récepteurs hormonaux positifs ou inconnus.
Traitement de deuxième ligne
ARIMIDEX 1 mg est indiqué pour le traitement du cancer du sein avancé chez les femmes ménopausées présentant une progression de la maladie après un traitement au tamoxifène. Les patients atteints d'une maladie ER-négative et les patients qui n'ont pas répondu à un traitement antérieur par le tamoxifène ont rarement répondu à ARIMIDEX.
DOSAGE ET ADMINISTRATION
Dose recommandée
La dose d'ARIMIDEX est d'un comprimé de 1 mg pris une fois par jour. Chez les patientes atteintes d'un cancer du sein avancé, ARIMIDEX 1 mg doit être poursuivi jusqu'à progression tumorale. ARIMIDEX peut être pris avec ou sans nourriture.
Pour le traitement adjuvant du cancer du sein précoce chez les femmes ménopausées, la durée optimale du traitement est inconnue. Dans l'essai ATAC, ARIMIDEX 1 mg a été administré pendant cinq ans [voir Etudes cliniques ].
Aucun ajustement posologique n'est nécessaire chez les patients insuffisants rénaux ou chez les patients âgés [voir Utilisation dans des populations spécifiques ].
Patients atteints d'insuffisance hépatique
Aucun changement de dose n'est recommandé chez les patients présentant une insuffisance hépatique légère à modérée. ARIMIDEX n'a pas été étudié chez les patients atteints d'insuffisance hépatique sévère [voir Utilisation dans des populations spécifiques ].
COMMENT FOURNIE
Formes posologiques et points forts
Les comprimés sont blancs, biconvexes, pelliculés contenant 1 mg d'anastrozole. Les comprimés sont imprimés d'un côté avec un logo composé d'une lettre « A » (majuscule) avec une pointe de flèche attachée au pied de la jambe droite étendue du « A » et au verso avec le marquage de force du comprimé « Adx 1 ”.
Stockage et manutention
Ces comprimés sont fournis en flacons de 30 comprimés ( CDN 0310-0201-30).
Stockage
Conserver à température ambiante contrôlée, 20-25°C (68-77°F) [voir USP ].
Distribué par : AstraZeneca Pharmaceuticals LP Wilmington, DE 19850. Révisé : Mai 2014
EFFETS SECONDAIRES
Les effets indésirables graves d'ARIMIDEX survenant chez moins de 1 patient sur 10 000 sont : 1) réactions cutanées telles que lésions, ulcères ou cloques ; 2) réactions allergiques avec gonflement du visage, des lèvres, de la langue et/ou de la gorge. Cela peut entraîner des difficultés à avaler et/ou à respirer ; et 3) modifications des tests sanguins de la fonction hépatique, y compris inflammation du foie accompagnée de symptômes pouvant inclure une sensation générale de malaise, avec ou sans ictère, douleur hépatique ou gonflement du foie.
Les effets indésirables fréquents (survenant avec une incidence ≥ 10 %) chez les femmes prenant ARIMIDEX 1 mg comprenaient : bouffées de chaleur, asthénie, arthrite, douleur, arthralgie, hypertension, dépression, nausées et vomissements, éruption cutanée, ostéoporose, fractures, douleurs dorsales, insomnie, maux de tête, douleurs osseuses, œdème périphérique, augmentation de la toux, dyspnée, pharyngite et lymphœdème.
Dans l'essai ATAC, l'effet indésirable signalé le plus fréquemment (> 0,1 %) entraînant l'arrêt du traitement dans les deux groupes de traitement était les bouffées de chaleur, bien que moins de patients aient arrêté le traitement en raison de bouffées de chaleur dans le groupe ARIMIDEX 1 mg.
Étant donné que les essais cliniques sont menés dans des conditions très variables, les taux d'effets indésirables observés dans les essais cliniques d'un médicament ne peuvent pas être directement comparés aux taux des essais cliniques d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique.
Expérience des essais cliniques
Thérapie adjuvante
Les données sur les effets indésirables du traitement adjuvant sont basées sur l'essai ATAC [voir Etudes cliniques ]. La durée médiane du traitement adjuvant pour l'évaluation de la sécurité était de 59,8 mois et 59,6 mois pour les patients recevant ARIMIDEX 1 mg et tamoxifène 20 mg, respectivement.
Les effets indésirables survenant avec une incidence d'au moins 5 % dans l'un ou l'autre des groupes de traitement pendant le traitement ou dans les 14 jours suivant la fin du traitement sont présentés dans le tableau 1.
Certains effets indésirables et combinaisons d'effets indésirables ont été spécifiés de manière prospective pour analyse, sur la base des propriétés pharmacologiques connues et des profils d'effets secondaires des deux médicaments (voir tableau 2).
Événements cardiovasculaires ischémiques
Entre les bras de traitement dans la population globale de 6186 patients, il n'y avait pas de différence statistique dans les événements cardiovasculaires ischémiques (ARIMIDEX 4 % contre tamoxifène 3 %).
Dans la population globale, une angine de poitrine a été rapportée chez 71/3092 (2,3%) patientes du bras ARIMIDEX 1mg et 51/3094 (1,6%) patientes du bras tamoxifène ; un infarctus du myocarde a été signalé chez 37/3092 (1,2 %) patients du bras ARIMIDEX 1 mg et 34/3094 (1,1 %) patients du bras tamoxifène.
Chez les femmes ayant une cardiopathie ischémique préexistante 465/6186 (7,5 %), l'incidence des événements cardiovasculaires ischémiques était de 17 % chez les patientes sous ARIMIDEX et de 10 % chez les patientes sous tamoxifène. Dans cette population de patients, une angine de poitrine a été signalée chez 25/216 (11,6 %) patients recevant ARIMIDEX et 13/249 (5,2 %) patients recevant du tamoxifène ; un infarctus du myocarde a été signalé chez 2 patients sur 216 (0,9 %) recevant ARIMIDEX et chez 8 patients sur 249 (3,2 %) recevant du tamoxifène.
Résultats de la densité minérale osseuse
Les résultats de la sous-étude osseuse de l'essai ATAC à 12 et 24 mois ont démontré que les patients recevant ARIMIDEX 1 mg présentaient une diminution moyenne de la densité minérale osseuse (DMO) de la colonne lombaire et de la hanche totale par rapport à la valeur initiale. Les patients recevant du tamoxifène présentaient une augmentation moyenne de la DMO de la colonne lombaire et de la hanche totale par rapport au départ.
Étant donné qu'ARIMIDEX abaisse les taux d'œstrogènes circulants, il peut entraîner une réduction de la densité minérale osseuse.
Un essai post-commercialisation a évalué les effets combinés d'ARIMIDEX 1 mg et du bisphosphonate risédronate sur les changements par rapport à la valeur initiale de la DMO et des marqueurs de la résorption et de la formation osseuses chez les femmes ménopausées atteintes d'un cancer du sein précoce à récepteurs hormonaux positifs. Tous les patients ont reçu une supplémentation en calcium et en vitamine D. À 12 mois, de petites réductions de la densité minérale osseuse du rachis lombaire ont été notées chez les patients ne recevant pas de bisphosphonates. Le traitement aux bisphosphonates a préservé la densité osseuse chez la plupart des patients à risque de fracture.
Les femmes ménopausées atteintes d'un cancer du sein précoce devant être traitées par ARIMIDEX 1 mg doivent voir leur état osseux géré conformément aux directives de traitement déjà disponibles pour les femmes ménopausées présentant un risque similaire de fracture de fragilité.
Cholestérol
Au cours de l'essai ATAC, un plus grand nombre de patients recevant ARIMIDEX 1 mg ont présenté un taux de cholestérol sérique élevé par rapport aux patients recevant du tamoxifène (9 % contre 3,5 %, respectivement).
Un essai post-commercialisation a également évalué les effets potentiels d'ARIMIDEX sur le profil lipidique. Dans la population d'analyse primaire pour les lipides (ARIMIDEX 1 mg seul), il n'y a eu aucun changement cliniquement significatif du LDL-C entre l'inclusion et 12 mois et du HDL-C entre l'inclusion et 12 mois.
Dans la population secondaire pour les lipides (ARIMIDEX + risédronate), il n'y a pas non plus eu de changement cliniquement significatif du LDL-C et du HDL-C entre l'inclusion et 12 mois.
Dans les deux populations pour les lipides, il n'y avait pas de différence cliniquement significative du cholestérol total (TC) ou des triglycérides sériques (TG) à 12 mois par rapport à la valeur initiale.
Dans cet essai, un traitement de 12 mois avec ARIMIDEX 1 mg seul a eu un effet neutre sur le profil lipidique. Le traitement combiné avec ARIMIDEX et le risédronate a également eu un effet neutre sur le profil lipidique.
L'essai fournit des preuves que les femmes ménopausées atteintes d'un cancer du sein précoce devant être traitées par ARIMIDEX 1 mg doivent être prises en charge conformément aux directives actuelles du programme national d'éducation sur le cholestérol pour la prise en charge basée sur le risque cardiovasculaire des patientes présentant des élévations de LDL.
Autres effets indésirables
Les patients recevant ARIMIDEX présentaient une augmentation des troubles articulaires (y compris l'arthrite, l'arthrose et l'arthralgie) par rapport aux patients recevant du tamoxifène. Les patients recevant ARIMIDEX présentaient une augmentation de l'incidence de toutes les fractures (en particulier les fractures de la colonne vertébrale, de la hanche et du poignet) [315 (10 %)] par rapport aux patients recevant du tamoxifène [209 (7 %)].
Les patients recevant ARIMIDEX 1 mg avaient une incidence plus élevée de syndrome du canal carpien [78 (2,5 %)] par rapport aux patients recevant du tamoxifène [22 (0,7 %)].
Les saignements vaginaux sont survenus plus fréquemment chez les patients traités au tamoxifène par rapport aux patients traités par ARIMIDEX 317 (10 %) contre 167 (5 %), respectivement.
Les patientes recevant ARIMIDEX présentaient une incidence plus faible de bouffées de chaleur, de saignements vaginaux, de pertes vaginales, de cancer de l'endomètre, d'événements thromboemboliques veineux et d'événements cérébrovasculaires ischémiques par rapport aux patientes recevant du tamoxifène.
Résultats médians d'innocuité du suivi sur 10 ans de l'essai ATAC
Les résultats sont cohérents avec les analyses précédentes.
Les effets indésirables graves étaient similaires entre ARIMIDEX (50 %) et le tamoxifène (51 %).
- Les événements cardiovasculaires correspondaient aux profils d'innocuité connus d'ARIMIDEX et du tamoxifène.
- L'incidence cumulée de toutes les premières fractures (graves et non graves, survenues pendant ou après le traitement) était plus élevée dans le groupe ARIMIDEX (15 %) que dans le groupe tamoxifène (11 %). Cette augmentation du taux de premières fractures pendant le traitement ne s'est pas poursuivie pendant la période de suivi post-traitement.
- L'incidence cumulée des nouveaux cancers primitifs était similaire dans le groupe ARIMIDEX 1 mg (13,7 %) par rapport au groupe tamoxifène (13,9 %). Conformément aux analyses précédentes, le cancer de l'endomètre était plus élevé dans le groupe tamoxifène (0,8 %) que dans le groupe ARIMIDEX (0,2 %).
- Le nombre total de décès (pendant ou hors essai) était similaire entre les groupes de traitement. Il y a eu plus de décès liés au cancer du sein dans le groupe tamoxifène que dans le groupe de traitement ARIMIDEX 1 mg.
Thérapie de première ligne
Les effets indésirables survenant avec une incidence d'au moins 5 % dans l'un ou l'autre des groupes de traitement des essais 0030 et 0027 pendant ou dans les 2 semaines suivant la fin du traitement sont présentés dans le tableau 3.
Les effets indésirables moins fréquents rapportés chez les patients recevant ARIMIDEX 1 mg l mg dans l'essai 0030 ou l'essai 0027 étaient similaires à ceux rapportés pour le traitement de deuxième intention.
Sur la base des résultats du traitement de deuxième ligne et du profil d'innocuité établi du tamoxifène, les incidences de 9 catégories d'événements indésirables pré-spécifiées potentiellement liées de manière causale à l'un ou aux deux traitements en raison de leur pharmacologie ont été analysées statistiquement. Aucune différence significative n'a été observée entre les groupes de traitement.
Thérapie de deuxième ligne
ARIMIDEX 1 mg a été toléré dans deux essais cliniques contrôlés (c'est-à-dire les essais 0004 et 0005), avec moins de 3,3 % des patients traités par ARIMIDEX et 4,0 % des patients traités par l'acétate de mégestrol se retirant en raison d'un effet indésirable.
Le principal effet indésirable plus fréquent avec ARIMIDEX qu'avec l'acétate de mégestrol était la diarrhée. Les effets indésirables rapportés chez plus de 5 % des patients dans l'un des groupes de traitement dans ces deux essais cliniques contrôlés, quelle que soit la causalité, sont présentés ci-dessous :
Corps dans son ensemble : syndrome grippal ; fièvre; la douleur du cou; malaise; blessure accidentelle; infection
Cardiovasculaire: Hypertension; thrombophlébite
Hépatique: Gamma GT augmenté ; SGOT augmenté ; augmentation de la SGPT Hématologie : anémie ; leucopénie
Métabolique et Nutritionnel : Phosphatase alcaline augmentée ; perte de poids
Les taux moyens de cholestérol total sérique ont augmenté de 0,5 mmol/L chez les patients recevant ARIMIDEX. Il a été démontré que les augmentations du cholestérol LDL contribuent à ces changements.
Musculo-squelettique : myalgie ; arthralgie; fracture pathologique
Nerveux: Somnolence; confusion; insomnie; anxiété; nervosité
Respiratoire: Sinusite; bronchite; rhinite
Peau et appendices : Amincissement des cheveux (alopécie); prurit
Urogénital: Infection urinaire; douleur mammaire
Les incidences des groupes d'effets indésirables suivants, potentiellement liés de manière causale à l'un ou aux deux traitements en raison de leur pharmacologie, ont été analysées statistiquement : prise de poids, œdème, maladie thromboembolique, troubles gastro-intestinaux, bouffées de chaleur et sécheresse vaginale. Ces six groupes, et les effets indésirables capturés dans les groupes, ont été définis de manière prospective. Les résultats sont présentés dans le tableau ci-dessous.
Expérience post-commercialisation
Ces effets indésirables sont signalés volontairement par une population de taille incertaine. Par conséquent, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale avec l'exposition aux médicaments. Les éléments suivants ont été signalés lors de l'utilisation post-approbation d'Arimidex :
- Événements hépatobiliaires, y compris augmentations de la phosphatase alcaline, de l'alanine aminotransférase, de l'aspartate aminotransférase, de la gamma-GT et de la bilirubine ; hépatite
- Éruption cutanée, y compris des cas de troubles cutanéo-muqueux tels que l'érythème polymorphe et le syndrome de Stevens-Johnson
- Cas de réactions allergiques incluant œdème de Quincke, urticaire et anaphylaxie [voir CONTRE-INDICATIONS ]
- Myalgie, doigt à ressaut et hypercalcémie (avec ou sans augmentation de l'hormone parathyroïdienne)
INTERACTIONS MÉDICAMENTEUSES
Tamoxifène
La co-administration d'anastrozole et de tamoxifène chez des patientes atteintes d'un cancer du sein a réduit la concentration plasmatique d'anastrozole de 27 %. Cependant, la co-administration d'anastrozole et de tamoxifène n'a pas affecté la pharmacocinétique du tamoxifène ou du N-desméthyltamoxifène. Au suivi médian de 33 mois, l'association d'ARIMIDEX et de tamoxifène n'a pas démontré de bénéfice d'efficacité par rapport au tamoxifène chez tous les patients ainsi que dans la sous-population positive aux récepteurs hormonaux. Ce bras de traitement a été retiré de l'essai [voir Etudes cliniques ]. D'après les résultats cliniques et pharmacocinétiques de l'essai ATAC, le tamoxifène ne doit pas être administré avec l'anastrozole.
Oestrogène
Les thérapies contenant des œstrogènes ne doivent pas être utilisées avec ARIMIDEX 1 mg car elles peuvent diminuer son action pharmacologique.
Warfarine
Dans une étude menée auprès de 16 hommes volontaires, l'anastrozole n'a pas modifié l'exposition (mesurée par la C max et l'ASC) ni l'activité anticoagulante (mesurée par le temps de prothrombine, le temps de thromboplastine partielle activée et le temps de thrombine) des R- et S- warfarine.
Cytochrome P450 Sur la base des résultats in vitro et in vivo, il est peu probable que la co-administration d'ARIMIDEX 1 mg affecte d'autres médicaments en raison de l'inhibition du cytochrome P450 [voir PHARMACOLOGIE CLINIQUE ].
AVERTISSEMENTS
Inclus dans le cadre du PRÉCAUTIONS section.
PRÉCAUTIONS
Événements cardiovasculaires ischémiques
Chez les femmes ayant une cardiopathie ischémique préexistante, une incidence accrue d'événements cardiovasculaires ischémiques a été observée avec ARIMIDEX dans l'essai ATAC (17 % des patientes sous ARIMIDEX 1 mg et 10 % des patientes sous tamoxifène). Tenir compte des risques et des avantages du traitement par ARIMIDEX chez les patients atteints de cardiopathie ischémique préexistante [voir EFFETS INDÉSIRABLES ]
Effets d'os
Les résultats de la sous-étude osseuse de l'essai ATAC à 12 et 24 mois ont démontré que les patients recevant ARIMIDEX présentaient une diminution moyenne de la densité minérale osseuse (DMO) de la colonne lombaire et de la hanche totale par rapport à la valeur initiale. Les patients recevant du tamoxifène présentaient une augmentation moyenne de la DMO de la colonne lombaire et de la hanche totale par rapport au départ. Envisager une surveillance de la densité minérale osseuse chez les patients traités par ARIMIDEX [voir EFFETS INDÉSIRABLES ].
Cholestérol
Au cours de l'essai ATAC, un plus grand nombre de patients recevant ARIMIDEX 1 mg présentaient une élévation du cholestérol sérique par rapport aux patients recevant du tamoxifène (9 % contre 3,5 %, respectivement) [voir EFFETS INDÉSIRABLES ].
Informations sur les conseils aux patients
Voir Étiquetage patient approuvé par la FDA (INFORMATIONS PATIENT).
Grossesse
Les patientes doivent être informées qu'ARIMIDEX 1 mg peut nuire au fœtus. Ils doivent également être informés qu'ARIMIDEX 1 mg ne doit pas être utilisé chez les femmes préménopausées ; par conséquent, si elles tombent enceintes, elles doivent arrêter de prendre ARIMIDEX et contacter immédiatement leur médecin.
Réactions allergiques (hypersensibilité)
Les patients doivent être informés de la possibilité de réactions allergiques graves avec gonflement du visage, des lèvres, de la langue et/ou de la gorge (œdème de Quincke) pouvant entraîner des difficultés à avaler et/ou à respirer et doivent consulter immédiatement un médecin.
Événements cardiovasculaires ischémiques
Les patients atteints d'une cardiopathie ischémique préexistante doivent être informés qu'une incidence accrue d'événements cardiovasculaires a été observée avec l'utilisation d'ARIMIDEX 1 mg par rapport à l'utilisation du tamoxifène. Si les patients ont des douleurs thoraciques nouvelles ou qui s'aggravent ou un essoufflement, ils doivent consulter immédiatement un médecin.
Effets d'os
Les patientes doivent être informées qu'ARIMIDEX abaisse le taux d'œstrogènes. Cela peut entraîner une perte de la teneur en minéraux des os, ce qui pourrait diminuer la solidité des os. Une conséquence possible de la diminution du contenu minéral des os est une augmentation du risque de fractures.
Cholestérol
Les patients doivent être informés qu'une augmentation du taux de cholestérol peut être observée lors du traitement par ARIMIDEX.
Chatouillement, fourmillement ou engourdissement
Les patients doivent être informés que s'ils ressentent des chatouillements, des picotements ou des engourdissements, ils doivent en informer leur fournisseur de soins de santé.
Tamoxifène
Les patients doivent être avisés de ne pas prendre ARIMIDEX avec du tamoxifène.
Doses oubliées
Informez les patients que s'ils oublient une dose, prenez-la dès qu'ils s'en souviennent. S'il est presque l'heure de leur prochaine dose, sautez la dose oubliée et prenez la prochaine dose régulièrement programmée. Les patients ne doivent pas prendre deux doses en même temps.
Toxicologie non clinique
Carcinogenèse, mutagenèse, altération de la fertilité
Une étude de carcinogenèse conventionnelle chez le rat à des doses de 1,0 à 25 mg/kg/jour (environ 10 à 243 fois la dose humaine quotidienne maximale recommandée sur une base en mg/m²) administrée par gavage oral pendant jusqu'à 2 ans a révélé une augmentation de la incidence des adénomes et des carcinomes hépatocellulaires et des polypes du stroma utérin chez les femelles et des adénomes thyroïdiens chez les mâles à la dose élevée. Une augmentation liée à la dose a été observée dans l'incidence de l'hyperplasie ovarienne et utérine chez les femelles. À 25 mg/kg/jour, les taux plasmatiques d'ASC0-24 h chez les rats étaient 110 à 125 fois plus élevés que le taux observé chez les volontaires ménopausées à la dose recommandée. Une étude distincte de cancérogénicité chez la souris à des doses orales de 5 à 50 mg/kg/jour (environ 24 à 243 fois la dose humaine quotidienne maximale recommandée sur une base en mg/m²) pendant jusqu'à 2 ans a produit une augmentation de l'incidence des affections bénignes. tumeurs des cellules ovariennes stromales, épithéliales et de la granulosa à toutes les doses. Une augmentation liée à la dose de l'incidence de l'hyperplasie ovarienne a également été observée chez les souris femelles. Ces changements ovariens sont considérés comme des effets spécifiques aux rongeurs de l'inhibition de l'aromatase et sont d'une importance discutable pour les humains. L'incidence de lymphosarcome a augmenté chez les mâles et les femelles à la dose élevée. À 50 mg/kg/jour, les taux plasmatiques d'ASC chez la souris étaient 35 à 40 fois plus élevés que le taux observé chez les volontaires ménopausées à la dose recommandée.
ARIMIDEX 1mg ne s'est pas révélé mutagène dans les tests in vitro (tests bactériens Ames et E. coli, test de mutation du gène CHO-K1) ou clastogène soit in vitro (aberrations chromosomiques dans les lymphocytes humains) soit in vivo (test du micronoyau chez le rat) .
L'administration orale d'anastrozole à des rats femelles (de 2 semaines avant l'accouplement au 7e jour de grossesse) a produit une incidence significative d'infertilité et réduit le nombre de grossesses viables à 1 mg/kg/jour (environ 10 fois la dose humaine recommandée en mg/m²). et 9 fois plus élevée que l'ASC0-24 h observée chez les volontaires ménopausées à la dose recommandée). La perte préimplantatoire d'ovules ou de fœtus a été augmentée à des doses égales ou supérieures à 0,02 mg/kg/jour (environ un cinquième de la dose humaine recommandée sur une base en mg/m²). Une récupération de la fertilité a été observée après une période sans administration de 5 semaines qui a suivi 3 semaines d'administration. On ne sait pas si ces effets observés chez les rats femelles sont révélateurs d'une altération de la fertilité chez l'homme.
Des études à doses multiples chez des rats ayant reçu de l'anastrozole pendant 6 mois à des doses égales ou supérieures à 1 mg/kg/jour (qui ont produit une Cssmax plasmatique et une ASC 0-24 h d'anastrozole 19 et 9 fois plus élevées que les valeurs respectives trouvées chez les femmes ménopausées volontaires à la dose recommandée) a entraîné une hypertrophie des ovaires et la présence de kystes folliculaires. De plus, des utérus hyperplasiques ont été observés dans des études de 6 mois chez des chiennes ayant reçu des doses égales ou supérieures à 1 mg/kg/jour (ce qui a produit une Cssmax plasmatique et une ASC0-24 h d'anastrozole 22 fois et 16 fois plus élevées que les doses respectives). valeurs trouvées chez les femmes ménopausées à la dose recommandée). On ne sait pas si ces effets sur les organes reproducteurs des animaux sont associés à une altération de la fertilité chez les femmes préménopausées.
Utilisation dans des populations spécifiques
Grossesse
Catégorie de grossesse X [voir CONTRE-INDICATIONS ]
ARIMIDEX peut nuire au fœtus lorsqu'il est administré à une femme enceinte et n'offre aucun avantage clinique aux femmes préménopausées atteintes d'un cancer du sein. ARIMIDEX est contre-indiqué chez les femmes enceintes ou susceptibles de l'être. Dans les études animales, l'anastrozole a provoqué un échec de grossesse, une augmentation des pertes de grossesse et des signes de retard de développement fœtal. Il n'y a pas d'études sur l'utilisation d'ARIMIDEX 1 mg chez les femmes enceintes. Si ARIMIDEX 1 mg est utilisé pendant la grossesse, ou si la patiente tombe enceinte pendant qu'elle reçoit ce médicament, la patiente doit être informée du danger potentiel pour le fœtus et du risque potentiel de perte de grossesse.
Dans les études de reproduction chez l'animal, des rats et des lapins gravides ont reçu de l'anastrozole pendant l'organogenèse à des doses égales ou supérieures à 1 (rats) et 1/3 (lapins) la dose humaine recommandée sur une base en mg/m². Chez les deux espèces, l'anastrozole a traversé le placenta et il y a eu une augmentation des pertes de grossesse (augmentation des pertes avant et/ou après l'implantation, augmentation de la résorption et diminution du nombre de fœtus vivants). Chez le rat, ces effets étaient liés à la dose et le poids du placenta était significativement augmenté. Une fœtotoxicité, y compris un développement fœtal retardé (c.-à-d. une ossification incomplète et une diminution du poids corporel du fœtus), s'est produite chez le rat à des doses d'anastrozole qui ont produit des concentrations plasmatiques maximales 19 fois plus élevées que les concentrations sériques chez l'homme à la dose thérapeutique (ASC 0-24 h 9 fois plus élevée) . Chez les lapines, l'anastrozole a provoqué un échec de grossesse à des doses égales ou supérieures à 16 fois la dose humaine recommandée sur une base en mg/m² [voir Toxicologie animale et/ou pharmacologie ].
Mères allaitantes
On ne sait pas si l'anastrozole est excrété dans le lait maternel. Étant donné que de nombreux médicaments sont excrétés dans le lait maternel et en raison de la tumorigénicité démontrée pour l'anastrozole dans les études animales, ou du risque d'effets indésirables graves chez les nourrissons, une décision doit être prise d'interrompre l'allaitement ou d'arrêter le médicament, en tenant compte de la importance du médicament pour la mère.
Utilisation pédiatrique
Les études cliniques chez les patients pédiatriques comprenaient un essai contrôlé par placebo chez des garçons pubères d'âge adolescent atteints de gynécomastie et un essai à un seul bras chez des filles atteintes du syndrome de McCune-Albright et d'une puberté précoce progressive. L'efficacité d'ARIMIDEX 1 mg dans le traitement de la gynécomastie pubertaire chez les garçons adolescents et dans le traitement de la puberté précoce chez les filles atteintes du syndrome de McCune-Albright n'a pas été démontrée.
Étude sur la gynécomastie
Une étude multicentrique randomisée, en double aveugle, contrôlée par placebo a recruté 80 garçons atteints de gynécomastie pubertaire âgés de 11 à 18 ans. Les patients ont été randomisés pour recevoir un régime quotidien d'ARIMIDEX 1 mg ou de placebo. Après 6 mois de traitement, il n'y avait pas de différence statistiquement significative dans le pourcentage de patients ayant présenté une réduction ≥ 50 % de la gynécomastie (analyse d'efficacité primaire). Les analyses d'efficacité secondaires (changement absolu du volume mammaire, pourcentage de patientes présentant une réduction du volume calculé de la gynécomastie, résolution de la douleur mammaire) étaient cohérentes avec l'analyse d'efficacité primaire. Les concentrations sériques d'estradiol au mois 6 de traitement ont été réduites de 15,4 % dans le groupe ARIMIDEX et de 4,5 % dans le groupe placebo.
Des effets indésirables évalués comme liés au traitement par les investigateurs sont survenus chez 16,3 % des patients traités par ARIMIDEX et 8,1 % des patients sous placebo, les plus fréquents étant l'acné (7 % ARIMIDEX et 2,7 % placebo) et les maux de tête (7 % ARIMIDEX 1mg et 0% placebo); tous les autres effets indésirables ont montré de petites différences entre les groupes de traitement. Un patient traité par ARIMIDEX 1 mg a interrompu l'essai en raison d'une hypertrophie testiculaire. La variation moyenne soustraite au départ du volume testiculaire après 6 mois de traitement était de + 6,6 ± 7,9 cm³ chez les patients traités par ARIMIDEX et de + 5,2 ± 8,0 cm³ dans le groupe placebo.
Étude sur le syndrome de McCune-Albright
Une étude multicentrique, à un seul bras et en ouvert a été menée auprès de 28 filles atteintes du syndrome de McCune-Albright et d'une puberté précoce progressive âgées de 2 à
Cinq patients (18 %) ont présenté des effets indésirables considérés comme possiblement liés à ARIMIDEX. Il s'agissait de nausées, d'acné, de douleurs dans un membre, d'une augmentation de l'alanine transaminase et de l'aspartate transaminase et d'une dermatite allergique.
Pharmacocinétique chez les patients pédiatriques
Après administration multiple de 1 mg une fois par jour chez des patients pédiatriques, le temps moyen pour atteindre la concentration maximale d'anastrozole était de 1 h. Les paramètres de disposition moyens (intervalle) de l'anastrozole chez les patients pédiatriques ont été décrits par un CL/F de 1,54 L/h (0,77-4,53 L/h) et un V/F de 98,4 L (50,7-330,0 L). La demi-vie d'élimination terminale était de 46,8 h, ce qui était similaire à celle observée chez les femmes ménopausées traitées par l'anastrozole pour un cancer du sein. D'après une analyse pharmacocinétique de population, la pharmacocinétique de l'anastrozole était similaire chez les garçons atteints de gynécomastie pubertaire et chez les filles atteintes du syndrome de McCune-Albright.
Utilisation gériatrique
Dans les études 0030 et 0027, environ 50 % des patients avaient 65 ans ou plus. Les patients âgés de ≥ 65 ans avaient une réponse tumorale et un délai de progression tumorale modérément meilleurs que les patients âgés de
Dans l'étude ATAC, 45 % des patients étaient âgés de 65 ans ou plus. L'efficacité d'ARIMIDEX 1mg par rapport au tamoxifène chez les patientes âgées de 65 ans ou plus (N=1413 pour ARIMIDEX et N=1410 pour le tamoxifène, le hazard ratio pour la survie sans maladie était de 0,93 [IC 95% : 0,80, 1,08]) était inférieure à l'efficacité observée chez les patientes de moins de 65 ans (N=1712 pour ARIMIDEX et N=1706 pour le tamoxifène, le hazard ratio pour la survie sans maladie était de 0,79 [IC 95% : 0,67, 0,94]).
La pharmacocinétique de l'anastrozole n'est pas affectée par l'âge.
Insuffisance rénale
Étant donné qu'environ 10 % seulement de l'anastrozole est excrété sous forme inchangée dans l'urine, l'insuffisance rénale n'influence pas la clairance corporelle totale. Une adaptation posologique chez les insuffisants rénaux n'est pas nécessaire [voir DOSAGE ET ADMINISTRATION et PHARMACOLOGIE CLINIQUE ].
Insuffisance hépatique
Les concentrations plasmatiques d'anastrozole chez les sujets atteints de cirrhose hépatique se situaient dans la plage des concentrations observées chez les sujets normaux dans tous les essais cliniques. Par conséquent, un ajustement posologique n'est pas non plus nécessaire chez les patients atteints de cirrhose hépatique stable. ARIMIDEX n'a pas été étudié chez les patients atteints d'insuffisance hépatique sévère [voir DOSAGE ET ADMINISTRATION et PHARMACOLOGIE CLINIQUE ].
SURDOSAGE
Des essais cliniques ont été menés avec ARIMIDEX, jusqu'à 60 mg en une seule dose administrée à des volontaires sains de sexe masculin et jusqu'à 10 mg par jour administrés à des femmes ménopausées atteintes d'un cancer du sein avancé ; ces doses ont été tolérées. Une dose unique d'ARIMIDEX 1 mg entraînant des symptômes potentiellement mortels n'a pas été établie. Il n'existe pas d'antidote spécifique en cas de surdosage et le traitement doit être symptomatique. Dans la gestion d'un surdosage, considérez que plusieurs agents peuvent avoir été pris. Des vomissements peuvent être provoqués si le patient est alerte. La dialyse peut être utile car ARIMIDEX n'est pas fortement lié aux protéines. Des soins de soutien généraux, y compris une surveillance fréquente des signes vitaux et une observation étroite du patient, sont indiqués.
CONTRE-INDICATIONS
Femmes enceintes et préménopausées
ARIMIDEX 1 mg peut être nocif pour le fœtus lorsqu'il est administré à une femme enceinte et n'offre aucun avantage clinique aux femmes préménopausées atteintes d'un cancer du sein. ARIMIDEX 1 mg est contre-indiqué chez les femmes enceintes ou susceptibles de l'être. Il n'y a pas d'études adéquates et bien contrôlées chez les femmes enceintes utilisant ARIMIDEX. Si ARIMIDEX 1 mg est utilisé pendant la grossesse, ou si la patiente tombe enceinte pendant qu'elle prend ce médicament, la patiente doit être informée du danger potentiel pour le fœtus ou du risque potentiel de perte de grossesse [voir Utilisation dans des populations spécifiques ].
Hypersensibilité
ARIMIDEX 1 mg est contre-indiqué chez tout patient ayant présenté une réaction d'hypersensibilité au médicament ou à l'un des excipients. Les réactions observées comprennent l'anaphylaxie, l'œdème de Quincke et l'urticaire [voir EFFETS INDÉSIRABLES ]
PHARMACOLOGIE CLINIQUE
Mécanisme d'action
La croissance de nombreux cancers du sein est stimulée ou entretenue par les œstrogènes.
Chez les femmes ménopausées, les œstrogènes proviennent principalement de l'action de l'enzyme aromatase, qui convertit les androgènes surrénaliens (principalement l'androstènedione et la testostérone) en œstrone et en œstradiol. La suppression de la biosynthèse des œstrogènes dans les tissus périphériques et dans le tissu cancéreux lui-même peut donc être obtenue en inhibant spécifiquement l'enzyme aromatase.
L'anastrozole est un inhibiteur sélectif non stéroïdien de l'aromatase. Il abaisse significativement les concentrations sériques d'œstradiol et n'a aucun effet détectable sur la formation de corticostéroïdes surrénaliens ou d'aldostérone.
Pharmacodynamie
Effet sur l'estradiol
Les concentrations sériques moyennes d'estradiol ont été évaluées dans des essais de dosage quotidiens multiples avec 0,5, 1, 3, 5 et 10 mg d'ARIMIDEX chez des femmes ménopausées atteintes d'un cancer du sein avancé. Une suppression cliniquement significative de l'œstradiol sérique a été observée avec toutes les doses. Des doses de 1 mg et plus ont entraîné la suppression des concentrations sériques moyennes d'estradiol jusqu'à la limite inférieure de détection (3,7 pmol/L). La dose quotidienne recommandée, ARIMIDEX 1 mg, a réduit l'estradiol d'environ 70 % en 24 heures et d'environ 80 % après 14 jours d'administration quotidienne. La suppression de l'œstradiol sérique a été maintenue jusqu'à 6 jours après l'arrêt de l'administration quotidienne d'ARIMIDEX 1 mg.
L'effet d'ARIMIDEX 1 mg chez les femmes préménopausées atteintes d'un cancer du sein précoce ou avancé n'a pas été étudié. Étant donné que l'aromatisation des androgènes surrénaliens n'est pas une source importante d'œstradiol chez les femmes préménopausées, ARIMIDEX ne devrait pas abaisser les taux d'œstradiol chez les femmes préménopausées.
Effet sur les corticostéroïdes
Dans plusieurs essais de dosage quotidien avec 3, 5 et 10 mg, la sélectivité de l'anastrozole a été évaluée en examinant les effets sur la synthèse des corticostéroïdes. Pour toutes les doses, l'anastrozole n'a pas affecté la sécrétion de cortisol ou d'aldostérone au départ ou en réponse à l'ACTH. Aucun traitement substitutif par glucocorticoïdes ou minéralocorticoïdes n'est nécessaire avec l'anastrozole.
Autres effets endocriniens
Dans plusieurs essais de dosage quotidien avec 5 et 10 mg, la thyréostimuline (TSH) a été mesurée; il n'y a pas eu d'augmentation de la TSH pendant l'administration d'ARIMIDEX. ARIMIDEX ne possède pas d'activité progestative, androgénique ou œstrogénique directe chez les animaux, mais perturbe les taux circulants de progestérone, d'androgènes et d'œstrogènes.
Pharmacocinétique
Absorption
L'inhibition de l'activité de l'aromatase est principalement due à l'anastrozole, le médicament parent. L'absorption de l'anastrozole est rapide et les concentrations plasmatiques maximales se produisent généralement dans les 2 heures suivant l'administration à jeun. Des études avec un médicament radiomarqué ont démontré que l'anastrozole administré par voie orale est bien absorbé dans la circulation systémique. La nourriture réduit le taux mais pas l'étendue globale de l'absorption de l'anastrozole. La C max moyenne de l'anastrozole a diminué de 16 % et le T max médian a été retardé de 2 à 5 heures lorsque l'anastrozole a été administré 30 minutes après un repas. La pharmacocinétique de l'anastrozole est linéaire sur la gamme de doses de 1 à 20 mg et ne change pas avec des doses répétées. La pharmacocinétique de l'anastrozole était similaire chez les patients et les volontaires sains.
Distribution
Les taux plasmatiques à l'état d'équilibre sont environ 3 à 4 fois plus élevés que les taux observés après une dose unique d'ARIMIDEX. Les concentrations plasmatiques approchent les niveaux de l'état d'équilibre après environ 7 jours d'administration une fois par jour. L'anastrozole est lié à 40 % aux protéines plasmatiques dans la plage thérapeutique.
Métabolisme
Le métabolisme de l'anastrozole se produit par N-désalkylation, hydroxylation et glucuronidation. Trois métabolites de l'anastrozole (le triazole, un conjugué glucuronide de l'hydroxy-anastrozole et un conjugué glucuronide de l'anastrozole lui-même) ont été identifiés dans le plasma et l'urine humains. Le principal métabolite circulant de l'anastrozole, le triazole, est dépourvu d'activité pharmacologique.
L'anastrozole a inhibé les réactions catalysées par le cytochrome P450 1A2, 2C8/9 et 3A4 in vitro avec des valeurs de Ki qui étaient environ 30 fois supérieures aux valeurs moyennes de Cmax à l'état d'équilibre observées après une dose quotidienne de 1 mg. L'anastrozole n'a eu aucun effet inhibiteur sur les réactions catalysées par le cytochrome P450 2A6 ou 2D6 in vitro. L'administration d'une dose unique de 30 mg/kg ou de doses multiples de 10 mg/kg d'anastrozole à des sujets sains n'a eu aucun effet sur la clairance de l'antipyrine ou sur la récupération urinaire des métabolites de l'antipyrine.
Excrétion
Quatre-vingt-cinq pour cent de l'anastrozole radiomarqué ont été récupérés dans les fèces et l'urine. Le métabolisme hépatique représente environ 85 % de l'élimination de l'anastrozole. L'élimination rénale représente environ 10 % de la clairance totale. La demi-vie d'élimination moyenne de l'anastrozole est de 50 heures.
Effet du sexe et de l'âge
La pharmacocinétique de l'anastrozole a été étudiée chez des femmes volontaires ménopausées et chez des patientes atteintes d'un cancer du sein. Aucun effet lié à l'âge n'a été observé entre 80 ans.
Effet de la race
Les taux sériques d'estradiol et de sulfate d'estrone étaient similaires entre les femmes ménopausées japonaises et caucasiennes qui ont reçu 1 mg d'anastrozole par jour pendant 16 jours. Les concentrations plasmatiques minimales moyennes à l'état d'équilibre de l'anastrozole chez les femmes ménopausées caucasiennes et japonaises étaient de 25,7 et 30,4 ng/mL, respectivement.
Effet de l'insuffisance rénale
La pharmacocinétique de l'anastrozole a été étudiée chez des sujets insuffisants rénaux. La clairance rénale de l'anastrozole a diminué proportionnellement à la clairance de la créatinine et était inférieure d'environ 50 % chez les volontaires présentant une insuffisance rénale sévère (clairance de la créatinine DOSAGE ET ADMINISTRATION et Utilisation dans des populations spécifiques ].
Effet de l'insuffisance hépatique
La pharmacocinétique de l'anastrozole a été étudiée chez des sujets atteints de cirrhose hépatique liée à l'abus d'alcool. La clairance orale apparente (CL/F) de l'anastrozole était environ 30 % plus faible chez les sujets atteints de cirrhose hépatique stable que chez les sujets témoins ayant une fonction hépatique normale. Cependant, ces concentrations plasmatiques restaient dans la fourchette des valeurs observées chez les sujets normaux. L'effet d'une insuffisance hépatique sévère n'a pas été étudié. Aucun ajustement posologique n'est nécessaire en cas de cirrhose hépatique stable [voir DOSAGE ET ADMINISTRATION et Utilisation dans des populations spécifiques ].
Toxicologie animale et/ou pharmacologie
Toxicologie reproductive
On a constaté que l'anastrozole traversait le placenta après administration orale de 0,1 mg/kg chez le rat et le lapin (environ 1 et 1,9 fois la dose humaine recommandée, respectivement, sur une base en mg/m²). Des études chez le rat et le lapin à des doses égales ou supérieures à 0,1 et 0,02 mg/kg/jour, respectivement (environ 1 et 1/3, respectivement, la dose humaine recommandée sur une base en mg/m²), administrées pendant la période de l'organogenèse a montré que l'anastrozole augmentait les pertes de grossesse (augmentation des pertes avant et/ou après l'implantation, augmentation de la résorption et diminution du nombre de fœtus vivants) ; les effets étaient liés à la dose chez le rat. Le poids du placenta a augmenté de façon significative chez les rats à des doses de 0,1 mg/kg/jour ou plus.
Des signes de fœtotoxicité, y compris un développement fœtal retardé (c.-à-d. une ossification incomplète et une diminution du poids corporel des fœtus), ont été observés chez des rats ayant reçu des doses de 1 mg/kg/jour (ce qui a produit une Cssmax plasmatique de l'anastrozole et une ASC 0-24 h qui étaient de 19 fois et 9 fois supérieures aux valeurs respectives trouvées chez les volontaires ménopausées à la dose recommandée). Il n'y avait aucune preuve de tératogénicité chez les rats ayant reçu des doses allant jusqu'à 1,0 mg/kg/jour. Chez les lapins, l'anastrozole a provoqué un échec de grossesse à des doses égales ou supérieures à 1,0 mg/kg/jour (environ 16 fois la dose humaine recommandée sur une base en mg/m²) ; il n'y avait aucune preuve de tératogénicité chez les lapins ayant reçu 0,2 mg/kg/jour (environ 3 fois la dose humaine recommandée sur une base en mg/m²).
Etudes cliniques
Traitement adjuvant du cancer du sein chez les femmes ménopausées
Un essai multicentrique en double aveugle (ATAC) a randomisé 9 366 femmes ménopausées atteintes d'un cancer du sein opérable pour un traitement adjuvant par ARIMIDEX 1 mg par jour, tamoxifène 20 mg par jour ou une combinaison des deux traitements pendant cinq ans ou jusqu'à la récidive de la maladie.
Le critère d'évaluation principal de l'essai était la survie sans maladie (c'est-à-dire le délai d'apparition d'une récidive distante ou locale, ou d'un cancer du sein controlatéral ou d'un décès quelle qu'en soit la cause). Les critères d'évaluation secondaires de l'essai comprenaient la survie sans maladie à distance, l'incidence du cancer du sein controlatéral et la survie globale. Au suivi médian de 33 mois, l'association d'ARIMIDEX et de tamoxifène n'a pas démontré de bénéfice d'efficacité par rapport au tamoxifène chez tous les patients ainsi que dans la sous-population positive aux récepteurs hormonaux. Ce bras de traitement a été retiré de l'essai. D'après les résultats cliniques et pharmacocinétiques de l'essai ATAC, le tamoxifène ne doit pas être administré avec l'anastrozole [voir INTERACTIONS MÉDICAMENTEUSES ].
Les caractéristiques démographiques et autres caractéristiques initiales étaient similaires dans les trois groupes de traitement (voir tableau 7).
Les patients des deux bras de monothérapie de l'essai ATAC ont été traités pendant une durée médiane de 60 mois (5 ans) et suivis pendant une durée médiane de 68 mois. La survie sans maladie dans la population en intention de traiter a été statistiquement significativement améliorée [Hazard Ratio (HR) = 0,87, IC à 95 % : 0,78, 0,97, p = 0,0127] dans le bras ARIMIDEX par rapport au bras tamoxifène. Dans la sous-population positive aux récepteurs hormonaux représentant environ 84 % des patientes de l'essai, la survie sans maladie a également été statistiquement significativement améliorée (HR = 0,83, IC à 95 % : 0,73, 0,94, p = 0,0049) dans le bras ARIMIDEX 1 mg par rapport au bras ARIMIDEX 1 mg. bras tamoxifène.
Figure 1 : Courbe de survie Kaplan Meier de survie sans maladie pour tous les patients randomisés pour recevoir ARIMIDEX 1 mg ou le tamoxifène en monothérapie dans l'essai ATAC (intention de traiter) 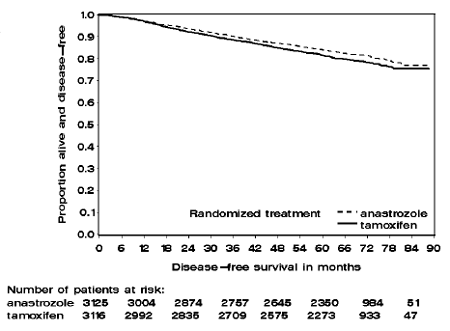
Figure 2 : Survie sans maladie pour la sous-population de patients positifs aux récepteurs hormonaux randomisés pour recevoir ARIMIDEX ou le tamoxifène en monothérapie dans l'essai ATAC 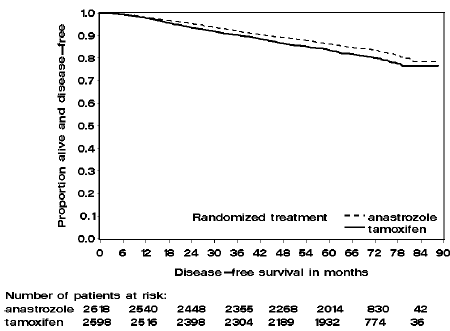
Les données de survie avec un suivi de 68 mois sont présentées dans le tableau 9.
Dans le groupe de patientes ayant déjà eu une chimiothérapie adjuvante (N=698 pour ARIMIDEX 1mg et N=647 pour le tamoxifène), le hazard ratio de survie sans maladie était de 0,91 (IC 95% : 0,73 à 1,13) dans le bras ARIMIDEX versus le bras tamoxifène.
La fréquence des événements individuels dans la population en intention de traiter et la sous-population positive aux récepteurs hormonaux est décrite dans le tableau 8.
Un résumé des résultats d'efficacité de l'étude est fourni dans le tableau 9.
Suivi médian sur 10 ans Efficacité Résultats de l'essai ATAC
Dans une analyse ultérieure de l'essai ATAC, les patients des deux bras de monothérapie ont été suivis pendant une durée médiane de 120 mois (10 ans). Les patients ont reçu le traitement de l'étude pendant une durée médiane de 60 mois (5 ans) (voir Tableau 10).
Figure 3 : Courbe de survie sans maladie de Kaplan Meier pour tous les patients randomisés pour recevoir ARIMIDEX ou le tamoxifène en monothérapie dans l'essai ATAC (intention de traiter)a 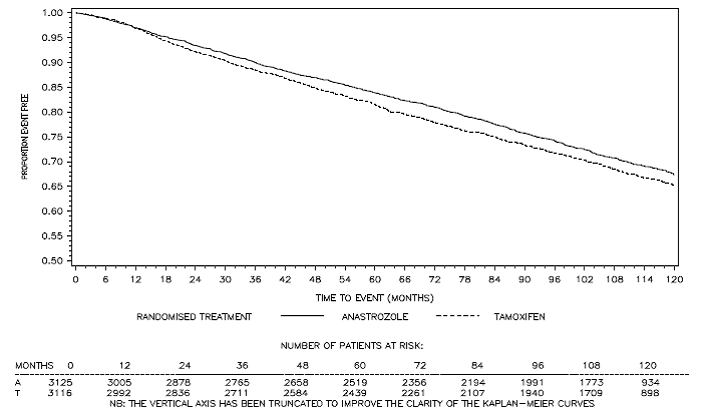
a La proportion de patients avec un suivi de 120 mois était de 29,4 %.
Figure 4 : Survie sans maladie pour la sous-population de patients positifs pour les récepteurs hormonaux randomisés pour recevoir ARIMIDEX 1 mg ou le tamoxifène en monothérapie dans l'essai ATACb 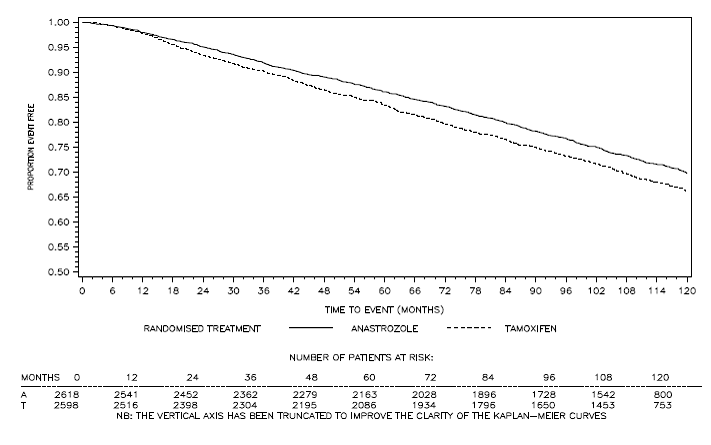
bLa proportion de patients avec un suivi de 120 mois était de 29,8 %.
Traitement de première intention chez les femmes ménopausées atteintes d'un cancer du sein avancé
Deux études cliniques contrôlées en double aveugle de conception similaire (0030, une étude nord-américaine et 0027, une étude à prédominance européenne) ont été menées pour évaluer l'efficacité d'ARIMIDEX 1 mg par rapport au tamoxifène en tant que traitement de première ligne pour les récepteurs hormonaux positifs ou les récepteurs hormonaux cancer du sein inconnu localement avancé ou métastatique chez les femmes ménopausées. Un total de 1021 patients âgés de 30 à 92 ans ont été randomisés pour recevoir le traitement d'essai. Les patients ont été randomisés pour recevoir 1 mg d'ARIMIDEX 1 mg une fois par jour ou 20 mg de tamoxifène une fois par jour. Les principaux critères d'évaluation des deux essais étaient le temps jusqu'à la progression tumorale, le taux de réponse objective de la tumeur et l'innocuité.
Les caractéristiques démographiques et autres caractéristiques de base, y compris les patients qui avaient une maladie mesurable et aucune maladie mesurable, les patients qui avaient reçu un traitement adjuvant antérieur, le site de la maladie métastatique et l'origine ethnique étaient similaires pour les deux groupes de traitement dans les deux essais. Le tableau suivant résume le statut des récepteurs hormonaux à l'entrée pour tous les patients randomisés dans les essais 0030 et 0027.
Pour les critères de jugement principaux, l'essai 0030 a montré qu'ARIMIDEX présentait un avantage statistiquement significatif par rapport au tamoxifène (p = 0,006) sur le délai de progression tumorale ; les taux de réponse tumorale objective étaient similaires pour ARIMIDEX 1 mg et le tamoxifène. L'essai 0027 a montré qu'ARIMIDEX 1 mg et le tamoxifène présentaient des taux de réponse tumorale objectifs et un délai de progression tumorale similaires (voir le tableau 12 et les figures 5 et 6).
Le tableau 12 ci-dessous résume les résultats de l'essai 0030 et de l'essai 0027 pour les critères principaux d'efficacité.
Figure 5 : Probabilité de Kaplan-Meier du temps jusqu'à la progression de la maladie pour tous les patients randomisés (en intention de traiter) dans l'essai 0030 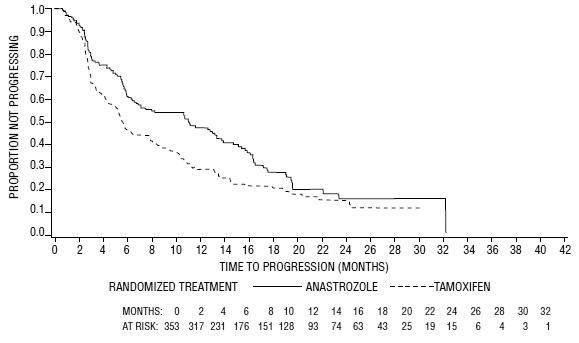
Figure 6 : Probabilité de Kaplan-Meier du temps jusqu'à progression pour tous les patients randomisés (intention de traiter) dans l'essai 0027 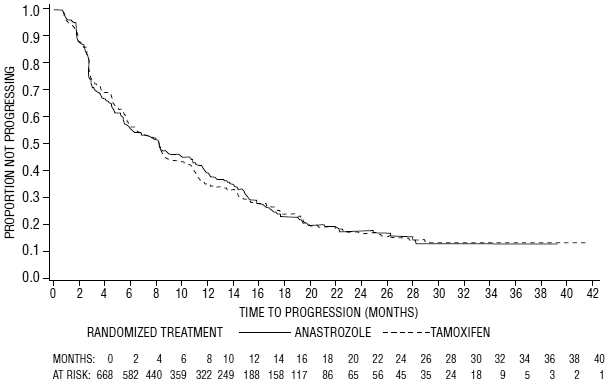
Les résultats des critères d'évaluation secondaires corroboraient les résultats des critères d'évaluation principaux de l'efficacité. Il y avait trop peu de décès survenus dans les groupes de traitement des deux essais pour tirer des conclusions sur les différences de survie globale.
Traitement de deuxième ligne chez les femmes ménopausées atteintes d'un cancer du sein avancé qui ont eu une progression de la maladie après un traitement au tamoxifène
L'anastrozole a été étudié dans deux essais cliniques contrôlés (0004, une étude nord-américaine ; 0005, une étude principalement européenne) chez des femmes ménopausées atteintes d'un cancer du sein avancé dont la maladie a progressé après un traitement au tamoxifène pour un cancer du sein avancé ou précoce. Certains des patients avaient également reçu un traitement cytotoxique antérieur. La plupart des patients étaient ER-positifs ; une plus petite fraction était ER-inconnu ou ER-négatif ; les patients ER-négatifs n'étaient éligibles que s'ils avaient eu une réponse positive au tamoxifène. Les patients éligibles atteints d'une maladie mesurable et non mesurable ont été randomisés pour recevoir soit une dose quotidienne unique de 1 mg, soit 10 mg d'ARIMIDEX, soit de l'acétate de mégestrol 40 mg quatre fois par jour. Les études étaient en double aveugle en ce qui concerne ARIMIDEX. Le temps jusqu'à la progression et les taux de réponse objective (seuls les patients atteints d'une maladie mesurable pouvaient être considérés comme des répondeurs partiels) étaient les principales variables d'efficacité. Les taux de réponse objective ont été calculés sur la base des critères de l'Union Internationale Contre le Cancer (UICC). Le taux de maladie stable prolongée (plus de 24 semaines), le taux de progression et la survie ont également été calculés.
Les deux essais ont inclus plus de 375 patients ; les données démographiques et les autres caractéristiques de base étaient similaires pour les trois groupes de traitement dans chaque essai. Les patients de l'essai 0005 avaient mieux répondu au traitement antérieur au tamoxifène. Parmi les patients inclus qui avaient déjà reçu un traitement au tamoxifène pour une maladie avancée (58 % dans l'essai 0004 ; 57 % dans l'essai 0005), 18 % de ces patients dans l'essai 0004 et 42 % dans l'essai 0005 ont été rapportés par l'investigateur principal comme ayant répondu. Dans l'essai 0004, 81 % des patients étaient ER-positifs, 13 % étaient ER-inconnus et 6 % étaient ER-négatifs. Dans l'essai 0005, 58 % des patients étaient ER-positifs, 37 % étaient ER-inconnus et 5 % étaient ER-négatifs. Dans l'essai 0004, 62 % des patients présentaient une maladie mesurable, contre 79 % dans l'essai 0005. Les sites de la maladie métastatique étaient similaires parmi les groupes de traitement pour chaque essai. En moyenne, 40 % des patients avaient des métastases des tissus mous ; 60 % avaient des métastases osseuses ; et 40 % avaient des métastases viscérales (15 % hépatiques).
Les résultats d'efficacité des deux études étaient similaires, comme présenté dans le tableau 13. Dans les deux études, il n'y avait aucune différence significative entre les bras de traitement en ce qui concerne l'un des paramètres d'efficacité répertoriés dans le tableau ci-dessous.
Lorsque les données des deux essais contrôlés sont regroupées, les taux de réponse objective et les délais médians jusqu'à la progression et le décès étaient similaires pour les patients randomisés pour recevoir ARIMIDEX 1 mg et l'acétate de mégestrol. Il n'y a, dans ces données, aucune indication qu'ARIMIDEX 10 mg soit supérieur à ARIMIDEX 1 mg.
INFORMATIONS PATIENTS
ARIMIDEX® A-rim-eh-dex (anastrozole) Comprimés pour administration orale
Quelles sont les informations les plus importantes que je devrais connaître sur ARIMIDEX ?
ARIMIDEX peut provoquer des effets secondaires graves, notamment :
- cardiopathie. Les femmes atteintes d'un cancer du sein à un stade précoce, qui ont des antécédents d'obstruction de leurs artères cardiaques (cardiopathie ischémique) et qui prennent ARIMIDEX 1 mg, peuvent présenter une augmentation des symptômes de diminution du flux sanguin vers leur cœur par rapport aux femmes similaires qui prennent du tamoxifène.
Consultez immédiatement un médecin si vous ressentez une douleur thoracique ou un essoufflement nouveau ou qui s'aggrave pendant le traitement par ARIMIDEX.
Qu'est-ce qu'ARIMIDEX ?
ARIMIDEX 1 mg est un médicament délivré sur ordonnance utilisé chez les femmes après la ménopause (« le changement de vie ») pour :
- traitement du cancer du sein précoce
- après l'opération
- chez les femmes dont le cancer du sein est positif aux récepteurs hormonaux
- le premier traitement du cancer du sein qui s'est propagé aux tissus voisins ou aux ganglions lymphatiques (localement avancé) ou qui s'est propagé à d'autres parties du corps (métastatique), chez les femmes dont le cancer du sein est positif aux récepteurs hormonaux ou dont les récepteurs hormonaux ne sont pas connus
- traitement du cancer du sein avancé, si le cancer s'est développé ou si la maladie s'est propagée après un traitement au tamoxifène
ARIMIDEX 1 mg n'agit pas chez les femmes atteintes d'un cancer du sein qui ne sont pas ménopausées (femmes préménopausées).
Qui ne devrait pas prendre ARIMIDEX ?
Ne prenez jamais ARIMIDEX 1 mg si vous :
- êtes enceinte ou susceptible de devenir enceinte. ARIMIDEX peut nuire à votre bébé à naître. Si vous tombez enceinte pendant que vous prenez ARIMIDEX, informez-en immédiatement votre médecin.
- n'ont pas traversé la ménopause (sont préménopausées)
- avez eu une réaction allergique grave à l'anastrozole ou à l'un des ingrédients d'ARIMIDEX. Voir la fin de cette notice pour la liste complète des ingrédients d'ARIMIDEX. Les symptômes d'une réaction allergique grave à ARIMIDEX comprennent : gonflement du visage, des lèvres, de la langue ou de la gorge, difficulté à respirer ou à avaler, urticaire et démangeaisons.
Que dois-je dire à mon médecin avant de prendre ARIMIDEX 1mg ?
Avant de prendre ARIMIDEX, informez votre médecin si vous :
- n'ont pas traversé la ménopause. Parlez-en à votre médecin si vous n'êtes pas sûr.
- avez ou avez eu un problème cardiaque
- on vous a dit que vous souffrez d'un amincissement ou d'une faiblesse osseuse (ostéoporose)
- avoir un taux de cholestérol élevé
- avoir d'autres conditions médicales
- êtes enceinte ou envisagez de devenir enceinte. ARIMIDEX peut nuire à votre bébé à naître. Voir « Qui ne devrait pas prendre ARIMIDEX ? »
- allaitez ou envisagez d'allaiter. On ne sait pas si ARIMIDEX passe dans le lait maternel. Vous et votre médecin devez décider si vous allez prendre ARIMIDEX 1 mg ou allaiter. Vous ne devriez pas faire les deux.
Informez votre médecin de tous les médicaments que vous prenez, y compris les médicaments sur ordonnance et en vente libre, les vitamines et les suppléments à base de plantes. Prévenez surtout votre médecin si vous prenez :
- tamoxifène. Vous ne devriez pas prendre ARIMIDEX si vous prenez du tamoxifène. La prise d'ARIMIDEX 1 mg avec du tamoxifène peut réduire la quantité d'ARIMIDEX 1 mg dans votre sang et peut empêcher ARIMIDEX 1 mg d'agir aussi bien.
- Médicaments contenant des œstrogènes. ARIMIDEX 1 mg peut ne pas fonctionner s'il est pris avec l'un de ces médicaments :
- la thérapie de remplacement d'hormone
- pilules contraceptives
- crèmes d'oestrogènes
- anneaux vaginaux
- suppositoires vaginaux
Connaissez les médicaments que vous prenez. Conservez-en une liste pour la montrer à votre médecin et à votre pharmacien lorsque vous recevez un nouveau médicament.
Comment dois-je prendre ARIMIDEX ?
- Prenez ARIMIDEX exactement comme votre médecin vous l'a dit.
- Continuez à prendre ARIMIDEX jusqu'à ce que votre médecin vous dise d'arrêter.
- ARIMIDEX 1 mg peut être pris avec ou sans nourriture.
- Si vous oubliez une dose, prenez-la dès que vous vous en souvenez. S'il est presque l'heure de votre prochaine dose, sautez la dose oubliée. Prenez votre prochaine dose régulière. Ne prenez pas deux doses en même temps.
Si vous avez pris trop d'ARIMIDEX 1 mg, appelez votre médecin ou rendez-vous immédiatement aux urgences de l'hôpital le plus proche.
Quels sont les effets secondaires possibles d'ARIMIDEX ?
ARIMIDEX peut provoquer des effets secondaires graves, notamment :
- Voir "Quelle est l'information la plus importante que je devrais connaître sur ARIMIDEX ?"
- amincissement ou faiblesse osseuse (ostéoporose). ARIMIDEX 1 mg réduit les œstrogènes dans votre corps, ce qui peut entraîner un amincissement et un affaiblissement de vos os. Cela peut augmenter votre risque de fractures, en particulier de la colonne vertébrale, de la hanche et du poignet. Votre médecin peut vous prescrire un test de densité minérale osseuse
- avant de commencer et pendant le traitement par ARIMIDEX pour vérifier si vous avez des modifications osseuses.
- augmentation du cholestérol sanguin (graisse dans le sang). Votre médecin peut effectuer des tests sanguins pour vérifier votre taux de cholestérol pendant que vous prenez ARIMIDEX.
- réactions cutanées. Arrêtez de prendre ARIMIDEX 1 mg et appelez immédiatement votre médecin si vous présentez des lésions cutanées, des ulcères ou des cloques.
- réactions allergiques sévères. Obtenez de l'aide médicale immédiatement si vous obtenez:
- gonflement du visage, des lèvres, de la langue ou de la gorge
- difficulté à avaler ou à respirer
- problèmes de foie. ARIMIDEX 1 mg peut provoquer une inflammation du foie et des modifications des tests sanguins de la fonction hépatique. Votre médecin peut vous vérifier pour cela. Arrêtez de prendre ARIMIDEX 1mg et appelez immédiatement votre médecin si vous présentez l'un de ces signes ou symptômes d'un problème de foie :
- un sentiment général de ne pas aller bien
- jaunissement de la peau ou du blanc des yeux
- douleur sur le côté droit de la région de l'estomac (abdomen)
Les effets secondaires courants chez les femmes prenant ARIMIDEX 1 mg comprennent :
- les bouffées de chaleur
- la faiblesse
- douleurs articulaires
- douleurs articulaires, raideur ou gonflement (arthrite)
- la douleur
- mal de gorge
- hypertension artérielle
- la dépression
- nausée et vomissements
- éruption
- mal au dos
- problèmes de sommeil
- douleur osseuse
- mal de tête
- gonflement des jambes, des chevilles ou des pieds
- augmentation de la toux
- essoufflement
- accumulation de liquide lymphatique dans les tissus de votre bras affecté (lymphœdème)
ARIMIDEX peut également provoquer des chatouillements, des picotements ou un engourdissement de la peau.
Dites à votre médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles d'ARIMIDEX. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
Comment dois-je conserver ARIMIDEX 1mg ?
- Conservez ARIMIDEX 1mg à température ambiante entre 68°F et 77°F (20°C à 25°C).
Gardez ARIMIDEX et tous les médicaments hors de la portée des enfants.
Informations générales sur l'utilisation sûre et efficace d'ARIMIDEX.
Les médicaments sont parfois prescrits à des fins autres que celles énumérées dans une notice d'information destinée aux patients. Ne prenez pas ARIMIDEX 1 mg pour une affection pour laquelle il n'a pas été prescrit. Ne donnez pas ARIMIDEX à d'autres personnes, même si elles présentent les mêmes symptômes que vous. Cela peut leur nuire.
Si vous souhaitez plus d'informations, parlez-en à votre médecin. Vous pouvez demander à votre pharmacien ou à votre médecin des informations sur ARIMIDEX destinées aux professionnels de la santé. Pour plus d'informations, appelez le 1-866-992-9276 ou visitez www.ARIMIDEX.com.
Quels sont les ingrédients d'ARIMIDEX ?
Ingrédient actif: anastrozole
Ingrédients inactifs: lactose, stéarate de magnésium, hydroxypropylméthylcellulose, polyéthylèneglycol, povidone, glycolate d'amidon sodique et dioxyde de titane.