Zovirax 200mg, 400mg, 800mg Acyclovir Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce que Zovirax 800 mg et comment est-il utilisé ?
Zovirax est un médicament d'ordonnance utilisé pour traiter les symptômes des boutons de fièvre (herpès labial) et de l'herpès génital. Zovirax 800 mg peut être utilisé seul ou avec d'autres médicaments.
Zovirax 400 mg appartient à une classe de médicaments appelés antiviraux topiques.
On ne sait pas si Zovirax 200 mg est sûr et efficace chez les enfants de moins de 12 ans.
Quels sont les effets secondaires possibles de Zovirax 800mg ?
Zovirax 200 mg peut provoquer des effets indésirables graves, notamment :
- ecchymoses ou saignements faciles,
- taches violettes ou rouges sous la peau,
- peu ou pas de miction,
- miction douloureuse ou difficile,
- gonflement des pieds ou des chevilles,
- se sentir fatigué, et
- essoufflement
Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires les plus courants de Zovirax incluent :
- nausée,
- vomissement,
- diarrhée,
- malaise général,
- maux de tête, et
- douleur buccale lors de l'utilisation d'un comprimé buccal d'acyclovir
Dites au médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles de Zovirax. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
LA DESCRIPTION
RÉSUMÉ DES INFORMATIONS SUR LE PRODUIT
Substance médicamenteuse
Nom propre: aciclovir
Nom chimique: 9-[(2-hydroxyéthoxy)méthyl]guanine
Autre nom: acycloguanosine
Formule moléculaire: C8H11N503
Masse moléculaire: 225.2 Formule développée :
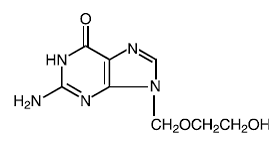
Propriétés physicochimiques : L'acyclovir est une poudre cristalline blanche dont la solubilité maximale dans l'eau est de 1,3 mg/mL à 25 °C.
LES INDICATIONS
Indications et utilisation clinique
ZOVIRAX® (acyclovir) est indiqué pour les conditions suivantes :
- Le traitement des premiers épisodes d'herpès génital.
- La suppression des récurrences inhabituellement fréquentes de l'herpès génital (6 épisodes ou plus par an).
- Le traitement aigu du zona (zona) et de la varicelle (varicelle).
Les résultats des études cliniques suggèrent que certains patients atteints d'herpès génital récurrent peuvent tirer un bénéfice clinique de l'administration de ZOVIRAX® par voie orale s'ils sont pris au premier signe d'un épisode imminent. Les patients les plus susceptibles d'en bénéficier sont les patients qui présentent des récidives sévères et prolongées ; un tel traitement intermittent peut être plus approprié qu'un traitement suppressif lorsque ces récidives sont peu fréquentes.
Le traitement précoce du zona aigu (zona) chez les personnes immunocompétentes avec ZOVIRAX® par voie orale a entraîné une diminution de l'excrétion virale ; diminution du temps de guérison ; moins de diffusion ; et le soulagement de la douleur aiguë.
Le traitement de la varicelle (varicelle) chez les patients immunocompétents avec ZOVIRAX® par voie orale a réduit le nombre total de lésions, accéléré la progression des lésions vers les stades croûteux et cicatrisés et diminué le nombre de lésions hypopigmentées résiduelles. De plus, ZOVIRAX® a diminué la fièvre et les symptômes constitutionnels associés à la varicelle.
L'utilisation prophylactique de l'acyclovir dans la varicelle n'a pas été établie.
Gériatrie (≥ 65 ans) : L'utilisation dans la population gériatrique peut être associée à des différences de sécurité dues à des modifications de la fonction rénale liées à l'âge et une brève discussion peut être trouvée dans les sections appropriées (voir AVERTISSEMENTS ET PRECAUTIONS ).
Pédiatrie ( Aucune donnée n'est disponible.
DOSAGE ET ADMINISTRATION
Considérations posologiques
- La posologie de ZOVIRAX® (acyclovir) doit être réduite chez les patients présentant une insuffisance rénale.
- Le traitement doit être initié dès que possible après un diagnostic de varicelle ou de zona, ou dès les premiers signes ou symptômes d'une poussée d'herpès génital.
- La dose recommandée et la durée d'utilisation dépendent de l'indication.
Dose recommandée et ajustement posologique
Traitement de l'infection initiale par l'herpès génital : 200 mg (un comprimé à 200 mg ou une cuillerée à thé de suspension [5 mL]) toutes les 4 heures, 5 fois par jour pour un total de 1 g par jour pendant 10 jours. Le traitement doit être instauré le plus tôt possible après l'apparition des signes et des symptômes.
Thérapie suppressive pour l'herpès génital récurrent
La dose initiale recommandée est de 200 mg (un comprimé de 200 mg ou une cuillerée à thé de suspension [5 mL]) trois fois par jour. Cela peut être augmenté si la percée se produit jusqu'à une dose d'un comprimé de 200 mg ou d'une cuillerée à thé [5 ml] de suspension, cinq fois par jour. Si nécessaire, une dose de 400 mg (deux comprimés de 200 mg ou deux cuillerées à thé de suspension [10 mL]) administrée deux fois par jour peut être envisagée. Une réévaluation périodique de la nécessité d'un traitement est recommandée.
L'administration de ZOVIRAX® pour le traitement intermittent est de 200 mg (un comprimé de 200 mg ou une cuillerée à thé [5 mL] de suspension) toutes les 4 heures 5 fois par jour pendant 5 jours. Le traitement doit être instauré dès le premier signe ou symptôme (prodrome) de récidive.
Traitement de l'herpès zoster
800 mg de ZOVIRAX® par voie orale, toutes les 4 heures, 5 fois par jour pendant 7 à 10 jours. Le traitement doit être instauré dans les 72 heures suivant l'apparition des lésions. Dans les essais cliniques, le plus grand bénéfice est survenu lorsque le traitement a été commencé dans les 48 heures suivant l'apparition des lésions.
Traitement de la varicelle
20 mg/kg (sans dépasser 800 mg) par voie orale, 4 fois par jour pendant 5 jours. Le traitement doit être instauré dans les 24 heures suivant l'apparition de l'éruption cutanée.
Patients atteints d'insuffisance rénale aiguë ou chronique
La prudence est recommandée lors de l'administration d'acyclovir à des patients présentant une insuffisance rénale. Une hydratation adéquate doit être maintenue.
Des études pharmacocinétiques complètes ont été réalisées après des perfusions intraveineuses d'acyclovir chez des patients insuffisants rénaux.
Sur la base de ces études, des ajustements posologiques sont recommandés dans le tableau 5 pour les indications d'herpès génital et de zona.
Hémodialyse
Pour les patients nécessitant une hémodialyse, la demi-vie plasmatique moyenne de l'acyclovir pendant l'hémodialyse est d'environ 5 heures. Il en résulte une diminution de 60 % des concentrations plasmatiques après une période de dialyse de six heures. Par conséquent, le schéma posologique du patient doit être ajusté de manière à ce qu'une dose supplémentaire soit administrée après chaque dialyse.
Dialyse péritonéale
Aucune dose supplémentaire ne semble nécessaire après ajustement de l'intervalle de dosage.
Dose oubliée
Si une dose de ZOVIRAX® est oubliée, le patient doit être avisé de la prendre dès qu'il s'en souvient, puis de continuer avec la dose suivante à l'intervalle de temps approprié.
COMMENT FOURNIE
Stockage et stabilité
Les comprimés ZOVIRAX® doivent être conservés à température ambiante contrôlée (15 à 25 °C) dans un endroit sec et à l'abri de la lumière.
La suspension ZOVIRAX® doit être conservée à température ambiante contrôlée (15 à 25 °C).
Formes posologiques, composition et conditionnement
Suspension: Chaque cuillerée à thé (5 ml) de suspension ZOVIRAX® contient 200 mg d'acyclovir et les ingrédients non médicinaux saveur de banane, cellulose, glycérine, méthylparabène, propylparabène, sorbitol, vanilline et eau.
Chaque comprimé ZOVIRAX® 200 contient 200 mg d'acyclovir et les ingrédients non médicinaux suivants : cellulose, indigotine, lactose, stéarate de magnésium, povidone et glycolate d'amidon sodique.
La suspension ZOVIRAX® est offerte en flacons de 125 mL* et de 475 mL. Chaque cuillerée à thé (5 ml) de suspension blanc cassé à saveur de banane contient 200 mg d'acyclovir.
*Bouteille de 125 ml non disponible au Canada
Les comprimés ZOVIRAX® 200 sont disponibles en flacons de 100 comprimés. Chaque comprimé bleu, en forme de bouclier, aux bords biseautés, contient 200 mg d'acyclovir et porte l'inscription « ZOVIRAX » sur une face et un triangle sur l'autre.
GlaxoSmithKline Inc., 7333 Mississauga Road, Mississauga, Ontario, L5N 6L4 1-800-387-7374. Révisé : 10 novembre 2014
EFFETS SECONDAIRES
Aperçu des effets indésirables du médicament
Les effets indésirables les plus fréquents associés à l'utilisation de ZOVIRAX® (acyclovir) sont les maux de tête et les nausées.
Des effets secondaires neurologiques ont également été signalés dans de rares cas. Les patients âgés et les patients ayant des antécédents d'insuffisance rénale présentent un risque accru de développer ces effets. Dans les cas rapportés, ces réactions étaient généralement réversibles à l'arrêt du traitement (voir AVERTISSEMENTS ET PRECAUTIONS et EFFETS INDÉSIRABLES , Effets indésirables post-commercialisation ).
Effets indésirables du médicament au cours des essais cliniques
Étant donné que les essais cliniques sont menés dans des conditions très spécifiques, les taux d'effets indésirables observés dans les essais cliniques peuvent ne pas refléter les taux observés dans la pratique et ne doivent pas être comparés aux taux des essais cliniques d'un autre médicament. Les informations sur les effets indésirables des médicaments issues des essais cliniques sont utiles pour identifier les événements indésirables liés aux médicaments et pour estimer les taux.
Traitement de l'herpès simplex
Administration à court terme (5 à 10 jours) : Les effets indésirables les plus fréquemment rapportés au cours des essais cliniques de traitement de l'herpès génital par ZOVIRAX® par voie orale chez 298 patients sont répertoriés dans le tableau 1.
Suppression de l'herpès simplex
Administration à long terme : Les événements indésirables les plus fréquemment rapportés dans un essai clinique pour la prévention des récidives avec l'administration continue de 400 mg (deux gélules de 200 mg) 2 fois par jour sont répertoriés dans le tableau 2.
Jusqu'à présent, les preuves issues des essais cliniques suggèrent que la gravité et la fréquence des événements indésirables ne nécessitent probablement pas l'arrêt du traitement.
Zona
Les effets indésirables les plus fréquemment rapportés au cours de trois essais cliniques de traitement du zona (zona) avec 800 mg de ZOVIRAX® par voie orale 5 fois par jour pendant 7 ou 10 jours ou un placebo sont répertoriés dans le tableau 3.
Varicelle
Les événements indésirables les plus fréquemment signalés au cours des trois essais cliniques de traitement de la varicelle par ZOVIRAX® oral ou un placebo sont énumérés dans le tableau 4.
Effets indésirables du médicament moins fréquents au cours des essais cliniques (
Les autres effets indésirables signalés chez moins de 1 % des patients recevant ZOVIRAX® dans le cadre d'un essai clinique comprenaient : douleur abdominale, anorexie, constipation, étourdissements, œdème, fatigue, flatulence, adénopathie inguinale, insomnie, douleur dans les jambes, goût des médicaments, éruption cutanée, gorge, mouvements spasmodiques des mains et urticaire.
Résultats hématologiques et biochimiques anormaux
Aucun changement cliniquement significatif des valeurs de laboratoire n'a été observé dans les essais cliniques pour le traitement de la varicelle et du zona, et pour le traitement et la suppression de l'herpès génital avec ZOVIRAX®.
Effets indésirables post-commercialisation
Les événements suivants ont été signalés volontairement lors de l'utilisation post-commercialisation de ZOVIRAX® dans la pratique clinique. Ces événements ont été choisis pour inclusion en raison de leur gravité, de leur fréquence de déclaration, de leur lien de causalité potentiel avec ZOVIRAX® ou d'une combinaison de ces facteurs. Les événements indésirables post-commercialisation sont signalés spontanément à partir d'une population de taille inconnue, il est donc impossible d'estimer la fréquence.
Général: Fièvre, maux de tête, douleurs et œdème périphérique.
Nerveux: Étourdissements, paresthésie, agitation, confusion, tremblements, ataxie, dysarthrie, hallucinations, symptômes psychotiques, convulsions, somnolence, encéphalopathie et coma ont été signalés. Ces événements sont généralement réversibles et généralement rapportés chez des patients insuffisants rénaux ou présentant d'autres facteurs prédisposants (voir AVERTISSEMENTS ET PRECAUTIONS ). Ces symptômes peuvent être marqués, en particulier chez les personnes âgées.
Digestif: Diarrhée, troubles gastro-intestinaux et nausées.
Hématologique et Lymphatique : Anémie, leucopénie, lymphadénopathie et thrombocytopénie.
Hypersensibilité et peau : Alopécie, érythème polymorphe, syndrome de Stevens-Johnson, nécrolyse épidermique toxique, éruptions cutanées incluant photosensibilité, prurit, urticaire, dyspnée, œdème de Quincke et anaphylaxie.
Tractus hépatobiliaire et pancréas : Rapports d'hyperbilirubinémie réversible et d'enzymes hépatiques élevées. Hépatite et jaunisse.
Musculo-squelettique : Myalgie.
Sens spéciaux : Anomalies visuelles.
Urogénital: Créatinine sanguine élevée et azote uréique sanguin (BUN). Une insuffisance rénale aiguë, des douleurs rénales et une hématurie ont été rapportées. La douleur rénale peut être associée à une insuffisance rénale (voir AVERTISSEMENTS ET PRECAUTIONS ).
INTERACTIONS MÉDICAMENTEUSES
Interactions médicament-médicament
Aucune interaction cliniquement significative n'a été identifiée.
L'acyclovir est éliminé principalement sous forme inchangée dans l'urine via une sécrétion tubulaire rénale active. Tous les médicaments administrés simultanément qui entrent en compétition avec ce mécanisme peuvent augmenter les concentrations plasmatiques d'acyclovir. Le probénécide et la cimétidine augmentent l'aire sous la courbe (ASC) de l'acyclovir par ce mécanisme et réduisent la clairance rénale de l'acyclovir. De même, des augmentations des ASC plasmatiques de l'acyclovir et du métabolite inactif du mycophénolate mofétil, un agent immunosuppresseur utilisé chez les patients transplantés, ont été observées lorsque les médicaments sont co-administrés. Cependant, aucun ajustement posologique n'est nécessaire en raison du large index thérapeutique de l'acyclovir.
Interactions médicament-aliment
Il n'y a pas d'interaction connue avec les aliments (voir Action et pharmacologie clinique , Pharmacocinétique ).
Interactions médicament-herbe
Les interactions avec les produits à base de plantes n'ont pas été établies.
Interactions médicament-test de laboratoire
Les interactions avec les tests de laboratoire n'ont pas été établies.
AVERTISSEMENTS
Les gélules, les comprimés et la suspension de ZOVIRAX (acyclovir) sont destinés à l'ingestion orale uniquement. Une insuffisance rénale, entraînant dans certains cas la mort, a été observée avec le traitement par l'acyclovir (voir EFFETS INDÉSIRABLES : observés pendant la pratique clinique et le SURDOSAGE ). Un purpura thrombotique thrombocytopénique/syndrome hémolytique et urémique (PTT/SHU), qui a entraîné la mort, est survenu chez des patients immunodéprimés recevant un traitement par acyclovir.
PRÉCAUTIONS
Un ajustement posologique est recommandé lors de l'administration de ZOVIRAX (acyclovir) à des patients insuffisants rénaux (voir DOSAGE ET ADMINISTRATION ). Il convient également d'être prudent lors de l'administration de ZOVIRAX (acyclovir) à des patients recevant des agents potentiellement néphrotoxiques car cela peut augmenter le risque de dysfonctionnement rénal et/ou le risque de symptômes réversibles du système nerveux central tels que ceux qui ont été rapportés chez les patients traités par acyclovir intraveineux. . Une hydratation adéquate doit être maintenue.
Zona: Il n'y a pas de données sur le traitement initié plus de 72 heures après le début de l'éruption de zona. Les patients doivent être informés qu'ils doivent commencer le traitement dès que possible après un diagnostic de zona.
Infections herpétiques génitales : Les patients doivent être informés que ZOVIRAX (acyclovir) ne guérit pas l'herpès génital. Il n'y a pas de données évaluant si ZOVIRAX (acyclovir) empêchera la transmission de l'infection à d'autres. L'herpès génital étant une maladie sexuellement transmissible, les patients doivent éviter tout contact avec des lésions ou des rapports sexuels lorsque des lésions et/ou des symptômes sont présents afin d'éviter d'infecter leurs partenaires. L'herpès génital peut également être transmis en l'absence de symptômes par excrétion virale asymptomatique. Si la prise en charge médicale d'une récidive d'herpès génital est indiquée, il faut conseiller aux patients de commencer le traitement dès le premier signe ou symptôme d'un épisode.
Varicelle: La varicelle chez les enfants par ailleurs en bonne santé est généralement une maladie auto-limitée de gravité légère à modérée. Les adolescents et les adultes ont tendance à avoir une maladie plus grave. Le traitement a été initié dans les 24 heures suivant l'éruption typique de la varicelle dans les études contrôlées, et il n'y a aucune information concernant les effets du traitement commencé plus tard dans l'évolution de la maladie.
Carcinogenèse, mutagenèse, altération de la fertilité
Les données présentées ci-dessous incluent des références aux concentrations plasmatiques maximales d'acyclovir à l'état d'équilibre observées chez les humains traités avec 800 mg administrés par voie orale 5 fois par jour (dosage approprié pour le traitement du zona) ou 200 mg administrés par voie orale 5 fois par jour (dosage approprié pour le traitement d'herpès génital). Les concentrations plasmatiques du médicament dans les études animales sont exprimées en multiples de l'exposition humaine à l'acyclovir aux schémas posologiques supérieurs et inférieurs (voir PHARMACOLOGIE CLINIQUE : Pharmacocinétique ).
L'acyclovir a été testé dans des essais biologiques à vie chez des rats et des souris à des doses quotidiennes uniques allant jusqu'à 450 mg/kg administrées par gavage. Il n'y avait pas de différence statistiquement significative dans l'incidence des tumeurs entre les animaux traités et les animaux témoins, et l'acyclovir n'a pas raccourci la latence des tumeurs. Les concentrations plasmatiques maximales étaient de 3 à 6 fois les niveaux humains dans le test biologique sur souris et de 1 à 2 fois les niveaux humains dans le test biologique sur rat.
L'acyclovir a été testé dans 16 tests de toxicité génétique in vitro et in vivo. L'acyclovir était positif dans 5 des tests.
L'acyclovir n'a pas altéré la fertilité ou la reproduction chez la souris (450 mg/kg/jour, po) ni chez le rat (25 mg/kg/jour, sc). Dans l'étude sur la souris, les niveaux plasmatiques étaient de 9 à 18 fois les niveaux humains, tandis que dans l'étude sur les rats, ils étaient de 8 à 15 fois les niveaux humains. À des doses plus élevées (50 mg/kg/jour, sc) chez le rat et le lapin (11 à 22 et 16 à 31 fois les concentrations humaines, respectivement), l'efficacité de l'implantation, mais pas la taille de la portée, a diminué. Dans une étude périnatale et postnatale chez le rat à 50 mg/kg/jour, sc, il y avait une diminution statistiquement significative du nombre moyen de groupe de corps jaunes, de sites d'implantation totaux et de fœtus vivants.
Aucune anomalie testiculaire n'a été observée chez les chiens ayant reçu 50 mg/kg/jour, IV pendant 1 mois (21 à 41 fois les niveaux humains) ou chez les chiens ayant reçu 60 mg/kg/jour par voie orale pendant 1 an (6 à 12 fois les niveaux humains). Une atrophie testiculaire et une aspermatogenèse ont été observées chez le rat et le chien à des doses plus élevées.
Grossesse
Effets tératogènes : Catégorie de grossesse B. L'acyc lovir administré pendant l'organogenèse n'a pas été tératogène chez la souris (450 mg/kg/jour, po), le lapin (50 mg/kg/jour, sc et IV) ou le rat (50 mg/kg/jour, sc). Ces expositions ont entraîné des niveaux plasmatiques de 9 et 18, 16 et 106, et 11 et 22 fois, respectivement, les niveaux humains.
Il n'y a pas d'études adéquates et bien contrôlées chez les femmes enceintes. Un registre épidémiologique prospectif de l'utilisation de l'acyclovir pendant la grossesse a été établi en 1984 et achevé en avril 1999. Il y a eu 749 grossesses suivies chez des femmes exposées à l'acyclovir systémique au cours du premier trimestre de la grossesse, entraînant 756 issues. Le taux d'occurrence des malformations congénitales se rapproche de celui observé dans la population générale. Cependant, la petite taille du registre est insuffisante pour évaluer le risque d'anomalies moins courantes ou pour permettre des conclusions fiables ou définitives concernant la sécurité de l'acyclovir chez les femmes enceintes et leurs fœtus en développement. L'acyclovir ne doit être utilisé pendant la grossesse que si le bénéfice potentiel justifie le risque potentiel pour le fœtus.
Mères allaitantes
Les concentrations d'acyclovir ont été documentées dans le lait maternel chez 2 femmes après l'administration orale de ZOVIRAX (acyclovir) et variaient de 0,6 à 4,1 fois les concentrations plasmatiques correspondantes. Ces concentrations exposeraient potentiellement le nourrisson à une dose d'acyclovir allant jusqu'à 0,3 mg/kg/jour. ZOVIRAX (acyclovir) doit être administré à une mère qui allaite avec prudence et uniquement lorsque cela est indiqué.
Utilisation pédiatrique
La sécurité et l'efficacité des formulations orales d'acyclovir chez les patients pédiatriques de moins de 2 ans n'ont pas été établies.
Utilisation gériatrique
Sur 376 sujets ayant reçu ZOVIRAX (acyclovir) dans une étude clinique sur le traitement du zona chez des sujets immunocompétents âgés de ¡Ý50 ans, 244 avaient 65 ans et plus tandis que 111 avaient 75 ans et plus. Aucune différence globale d'efficacité pour le temps jusqu'à l'arrêt de la formation de nouvelles lésions ou le temps de guérison n'a été signalée entre les sujets gériatriques et les sujets adultes plus jeunes. La durée de la douleur après cicatrisation était plus longue chez les patients de 65 ans et plus. Nausées, vomissements et étourdissements ont été signalés plus fréquemment chez les sujets âgés. Les patients âgés sont plus susceptibles d'avoir une fonction rénale réduite et nécessitent une réduction de la dose. Les patients âgés sont également plus susceptibles d'avoir des événements indésirables rénaux ou du système nerveux central. En ce qui concerne les effets indésirables sur le SNC observés au cours de la pratique clinique, la somnolence, les hallucinations, la confusion et le coma ont été signalés plus fréquemment chez les patients âgés (voir PHARMACOLOGIE CLINIQUE, EFFETS INDÉSIRABLES : Observés au cours de la pratique clinique, et DOSAGE ET ADMINISTRATION ).
SURDOSAGE
Pour la prise en charge d'une surdose suspectée de drogue, contactez votre centre antipoison régional.
Du charbon actif peut être administré pour faciliter l'élimination du médicament non absorbé. Des mesures générales de soutien sont recommandées.
L'acyclovir n'est que partiellement absorbé dans le tractus gastro-intestinal. Les patients ont ingéré jusqu'à 20 g d'acyclovir en une seule occasion, sans effets indésirables inattendus. Dans les études cliniques, la concentration plasmatique la plus élevée observée chez un seul patient à ces doses était de 10,0 μg/mL. Des surdosages accidentels et répétés d'acyclovir oral sur plusieurs jours ont été associés à des effets gastro-intestinaux (tels que nausées et vomissements) et neurologiques (maux de tête et confusion).
Les doses intraveineuses administrées à l'homme ont été aussi élevées que 1 200 mg/m² (28 mg/kg) 3 fois par jour pendant jusqu'à 2 semaines. Les concentrations plasmatiques maximales ont atteint 80 μg/mL. Le surdosage d'acyclovir intraveineux a entraîné des élévations de la créatinine sérique, de l'azote uréique du sang et une insuffisance rénale subséquente. Des effets neurologiques incluant confusion, hallucinations, agitation, convulsions et coma ont été décrits en association avec un surdosage par voie intraveineuse.
Les patients doivent être étroitement surveillés afin de déceler tout signe de toxicité. L'hémodialyse améliore significativement l'élimination de l'acyclovir du sang et peut donc être considérée comme une option de prise en charge en cas de surdosage symptomatique. Une précipitation de l'acyclovir dans les tubules rénaux peut se produire si la solubilité (2,5 mg/mL) dans le liquide intratubulaire est dépassée. En cas d'insuffisance rénale et d'anurie, le patient peut bénéficier d'une hémodialyse jusqu'au rétablissement de la fonction rénale (voir DOSAGE ET ADMINISTRATION ).
CONTRE-INDICATIONS
ZOVIRAX® (acyclovir) est contre-indiqué chez les patients qui développent une hypersensibilité ou qui sont hypersensibles à l'acyclovir, au valacyclovir ou à tout autre composant des formulations de ZOVIRAX®. Pour une liste complète, voir Formes posologiques , Composition et Conditionnement section de la monographie du produit .
PHARMACOLOGIE CLINIQUE
Action et pharmacologie clinique
Mécanisme d'action
ZOVIRAX® (acyclovir), un analogue nucléosidique purique acyclique synthétique, est un substrat avec un haut degré de spécificité pour l'herpès simplex et la thymidine kinase spécifiée pour la varicelle-zona. L'acyclovir est un mauvais substrat pour la thymidine kinase spécifiée par la cellule hôte. La thymidine kinase spécifiée par l'herpès simplex et la varicelle-zona transforme l'acyclovir en son monophosphate qui est ensuite transformé par un certain nombre d'enzymes cellulaires en acyclovir diphosphate et acyclovir triphosphate. L'acyclovir triphosphate est à la fois un inhibiteur et un substrat de l'ADN polymérase spécifiée par l'herpèsvirus. Bien que l'α-ADN polymérase cellulaire dans les cellules infectées puisse également être inhibée par l'acyclovir triphosphate, cela ne se produit qu'à des concentrations d'acyclovir triphosphate qui sont supérieures à celles qui inhibent l'ADN polymérase spécifiée par l'herpèsvirus. L'acyclovir est sélectivement converti en sa forme active dans les cellules infectées par l'herpèsvirus et est ainsi préférentiellement capté par ces cellules. L'acyclovir a démontré un potentiel toxique très inférieur in vitro pour les cellules normales non infectées parce que : 1) moins est absorbé ; 2) less est converti en la forme active ; et 3) l'α-ADN polymérase cellulaire a une sensibilité plus faible à l'action de la forme active du médicament. Une combinaison de la spécificité de la thymidine kinase, de l'inhibition de l'ADN polymérase et de l'arrêt prématuré de la synthèse de l'ADN entraîne l'inhibition de la réplication du virus de l'herpès. Aucun effet sur les virus latents non réplicatifs n'a été démontré. L'inhibition du virus réduit la période d'excrétion virale, limite le degré de propagation et le niveau de pathologie et facilite ainsi la guérison. Pendant la suppression, il n'y a aucune preuve que l'acyclovir empêche la migration neuronale du virus. Il interrompt les épisodes d'herpès récurrents en raison de l'inhibition de la réplication virale après la réactivation.
Pharmacocinétique
La pharmacocinétique de l'acyclovir après administration orale a été évaluée dans 6 études cliniques portant sur 110 patients adultes.
Absorption
Dans une étude portant sur 35 patients immunodéprimés atteints d'herpès simplex ou d'infection varicelle-zona ayant reçu des capsules ZOVIRAX® à des doses de 200 à 1 000 mg toutes les 4 heures, 6 fois par jour pendant 5 jours, la biodisponibilité a été estimée à 15 à 20 %. Dans cette étude, les concentrations plasmatiques à l'état d'équilibre ont été atteintes dès le deuxième jour d'administration. Les concentrations maximales et minimales moyennes à l'état d'équilibre après la dernière dose de 200 mg étaient de 0,49 μg/mL (0,47 à 0,54 μg/mL) et de 0,31 μg/mL (0,18 à 0,41 μg/mL), respectivement et après la dernière dose de 800 mg étaient 2,8 μg/mL (2,3 à 3,1 μg/mL) et 1,8 μg/mL (1,3 à 2,5 μg/mL). Dans une autre étude, 20 patients immunocompétents atteints d'infections génitales récurrentes à herpès simplex ont reçu des capsules de ZOVIRAX® à raison de 800 mg toutes les 6 heures, 4 fois par jour pendant 5 jours, les concentrations moyennes de pic et de creux à l'état d'équilibre étaient de 1,4 μg/mL (0,66 à 1,8 μg/mL) et 0,55 μg/mL (0,14 à 1,1 μg/mL).
Dans une étude croisée à doses multiples au cours de laquelle 23 volontaires ont reçu ZOVIRAX® sous forme d'une capsule de 200 mg, d'un comprimé de 400 mg et d'un comprimé de 800 mg 6 fois par jour, l'absorption a diminué avec l'augmentation de la dose et les biodisponibilités estimées de l'acyclovir étaient de 20, 15 et 10 %. , respectivement. On pense que la diminution de la biodisponibilité est fonction de la dose et non de la forme posologique. Il a été démontré que l'acyclovir n'est pas proportionnel à la dose sur la plage de dosage de 200 à 800 mg. Dans cette étude, les concentrations maximales et minimales d'acyclovir à l'état d'équilibre étaient de 0,83 et 0,46 μg/mL, 1,21 et 0,63 μg/mL et 1,61 et 0,83 μg/mL pour les schémas posologiques de 200, 400 et 800 mg, respectivement.
Dans une autre étude chez 6 volontaires, l'influence de la nourriture sur l'absorption de l'acyclovir n'était pas apparente.
Une étude de biodisponibilité d'une dose orale unique chez 23 volontaires sains a montré que les capsules ZOVIRAX® 200 mg sont bioéquivalentes à 200 mg d'acyclovir en solution aqueuse. Dans une étude distincte menée auprès de 20 volontaires, il a été démontré que la suspension ZOVIRAX® est bioéquivalente aux capsules ZOVIRAX®. Dans une autre étude de biodisponibilité/bioéquivalence à dose unique menée auprès de 24 volontaires, il a été démontré qu'un comprimé de ZOVIRAX® à 800 mg est bioéquivalent à quatre capsules de ZOVIRAX® à 200 mg.
Distribution
La liaison aux protéines plasmatiques est relativement faible (9 à 33 %) et les interactions médicamenteuses impliquant un déplacement du site de liaison ne sont pas anticipées.
Élimination
Après administration orale, la demi-vie plasmatique moyenne de l'acyclovir chez les volontaires et les patients ayant une fonction rénale normale variait de 2,5 à 3,3 heures. L'excrétion rénale moyenne du médicament inchangé représente 14,4 % (8,6 à 19,8 %) de la dose administrée par voie orale. Le seul métabolite urinaire (identifié par chromatographie liquide à haute performance) est la 9-[(carboxyméthoxy)méthyl]guanine.
Populations et conditions particulières
Pédiatrie
En général, la pharmacocinétique de l'acyclovir chez les enfants est similaire à celle des adultes. La demi-vie moyenne après des doses orales de 300 et 600 mg/m², chez les enfants âgés de 7 mois à 7 ans, était de 2,6 heures (intervalle de 1,59 à 3,74 heures).
L'acyclovir administré par voie orale chez les enfants de moins de 2 ans n'a pas encore été complètement étudié.
Gériatrie
Chez les personnes âgées, la clairance corporelle totale diminue avec l'âge, associée à une diminution de la clairance de la créatinine, bien qu'il y ait peu de changement dans la demi-vie plasmatique terminale. Une réduction de la posologie peut être nécessaire chez les patients gériatriques dont la fonction rénale est réduite (voir DOSAGE ET ADMINISTRATION ).
Insuffisance rénale
La demi-vie et la clairance corporelle totale de l'acyclovir dépendent de la fonction rénale.
Un ajustement posologique est recommandé chez les patients dont la fonction rénale est réduite (voir DOSAGE ET ADMINISTRATION ).
Essais cliniques
Herpès génital initial
Des études en double aveugle contrôlées par placebo ont démontré que ZOVIRAX® administré par voie orale réduisait significativement la durée de l'infection aiguë et la durée de la cicatrisation des lésions. La durée de la douleur et de la formation de nouvelles lésions a été réduite dans certains groupes de patients.
Herpès génital récurrent
Dans une étude portant sur des patients ayant reçu ZOVIRAX® à 400 mg deux fois par jour pendant 3 ans, 45, 52 et 63 % des patients sont restés sans récidive au cours de la première, de la deuxième et de la troisième années, respectivement. Des analyses en série des taux de récidive à 3 mois pour les patients ont montré que 71 à 87 % étaient sans récidive à chaque trimestre.
Infections à l'herpès zoster
Dans une étude en double aveugle contrôlée par placebo menée auprès de patients immunocompétents atteints d'un zona cutané localisé, ZOVIRAX® (800 mg 5 fois par jour pendant 10 jours) a réduit les délais d'apparition de la formation de croûtes, de cicatrisation et d'arrêt complet de la douleur, et a réduit la durée de l'infection virale. excrétion et la durée de la formation de nouvelles lésions.
Dans une étude similaire en double aveugle contrôlée par placebo, ZOVIRAX® (800 mg 5 fois par jour pendant 7 jours) a réduit les délais d'achèvement de la formation de la croûte, de la cicatrisation et de l'arrêt de la douleur, et a réduit la durée de la formation de nouvelles lésions.
Le traitement a été commencé dans les 72 heures suivant l'apparition de l'éruption cutanée et était plus efficace s'il était commencé dans les 48 premières heures. Les adultes de plus de 50 ans ont montré un plus grand bénéfice.
Varicelle
Trois essais randomisés, en double aveugle et contrôlés par placebo ont été menés chez 993 patients pédiatriques âgés de 2 à 18 ans atteints de varicelle. Tous les patients ont été traités dans les 24 heures suivant le début de l'éruption cutanée. Dans deux essais, ZOVIRAX® a été administré à raison de 20 mg/kg quatre fois par jour (jusqu'à 3 200 mg par jour) pendant 5 jours. Dans le troisième essai, des doses de 10, 15 ou 20 mg/kg ont été administrées quatre fois par jour pendant 5 à 7 jours. Le traitement par ZOVIRAX® a raccourci le temps de guérison à 50 %, réduit le nombre maximal de lésions, réduit le nombre médian de vésicules, diminué le nombre médian de lésions résiduelles au jour 28 et diminué la proportion de patients souffrant de fièvre, d'anorexie et de léthargie au jour 2. Le traitement par ZOVIRAX® n'a pas affecté les réponses immunitaires humorales ou cellulaires spécifiques au virus varicelle-zona 1 mois ou 1 an après le traitement.
Pharmacologie détaillée
Voir Action et pharmacologie clinique.
Virologie
La relation quantitative entre la sensibilité in vitro du virus de l'herpès simplex (HSV) et du virus varicelle-zona (VZV) à l'acyclovir et la réponse clinique au traitement n'a pas été établie chez l'homme, et les tests de sensibilité au virus n'ont pas été standardisés. Les résultats des tests de sensibilité, exprimés en tant que concentration de médicament nécessaire pour inhiber de 50 % la croissance du virus dans la culture cellulaire (ID50), varient considérablement en fonction du test particulier utilisé, du type de cellule utilisé et du laboratoire effectuant le test. La DI50 de l'acyclovir contre les isolats de HSV-1 peut aller de 0,02 μg/mL (réduction de la plaque dans les cellules Vero) à 5,9-13,5 μg/mL (réduction de la plaque dans les cellules de rein de singe vert [GMK]). L'ID50 contre HSV-2 varie de 0,01 à 9,9 μg/mL (réduction de la plaque dans les cellules Vero et GMK, respectivement).
En utilisant une méthode d'absorption de colorant dans des cellules Vero, qui donne des valeurs ID50 environ 5 à 10 fois plus élevées que les tests de réduction de plaque, 1 417 isolats de HSV (553 HSV-1 et 864 HSV-2) d'environ 500 patients ont été examinés sur une période de 5 période de l'année. Ces tests ont révélé que 90 % des isolats de HSV-1 étaient sensibles à ≤ 0,9 μg/mL d'acyclovir et 50 % de tous les isolats étaient sensibles à ≤ 0,2 μg/mL d'acyclovir. Pour les isolats HSV-2, 90 % étaient sensibles à ≤ 2,2 μg/mL et 50 % de tous les isolats étaient sensibles à ≤ 0,7 μg/mL d'acyclovir. Des isolats avec une sensibilité significativement diminuée ont été trouvés chez 44 patients. Il faut souligner que ni les patients ni les isolats n'ont été sélectionnés au hasard et, par conséquent, ne représentent pas la population générale. La plupart des isolats cliniques de HSV les moins sensibles ont été relativement déficients en thymidine kinase virale (TK). Des souches présentant des altérations de la TK virale ou de l'ADN polymérase virale ont également été signalées.
L'ID50 contre le VZV varie de 0,17 à 1,53 μg/mL (réduction du rendement, fibroblastes de prépuce humain) à 1,85 à 3,98 μg/mL (réduction des foyers, fibroblastes d'embryon humain [HEF]). La reproduction du génome de l'EBV est supprimée de 50 % dans les cellules Raji surinfectées ou les cellules lymphoblastoïdes P3HR-1 par 1,5 μg/mL d'acyclovir. Le cytomégalovirus (CMV) est relativement résistant à l'acyclovir avec des valeurs ID50 allant de 2,3-17,6 μg/mL (réduction de plaque, cellules HEF) à 1,82-56,8 μg/mL (hybridation ADN, cellules HEF). L'état latent du génome de l'un des virus de l'herpès humain n'est pas connu pour être sensible à l'acyclovir.
La résistance
L'exposition prolongée du HSV à des concentrations sous-inhibitrices (0,1 μg/mL) d'acyclovir en culture cellulaire a entraîné l'émergence d'une variété de souches résistantes à l'acyclovir. On pense que l'émergence de souches résistantes se produit par « sélection » de virus naturels présentant une sensibilité relativement faible à l'acyclovir. De telles souches ont été signalées dans des isolats pré-thérapeutiques issus de plusieurs études cliniques.
Deux mécanismes de résistance impliquant la thymidine kinase virale (nécessaire à l'activation de l'acyclovir) ont été décrits. Ce sont : (a) la sélection de mutants déficients en thymidine-kinase qui induisent peu ou pas d'activité enzymatique après l'infection, et (b) la sélection de mutants possédant une thymidine-kinase de spécificité de substrat altérée qui est capable de phosphoryler le nucléoside naturel thymidine mais pas aciclovir. La majorité des virus moins sensibles apparaissant in vitro sont du type déficient en thymidine-kinase qui ont une infectiosité et une pathogénicité réduites et une moindre probabilité d'induire une latence chez les animaux.
Cependant, une infection à HSV résistante à l'acyclovir chez un greffé de moelle osseuse immunodéprimé sous traitement prolongé à l'acyclovir s'est avérée être due à un isolat clinique qui avait une thymidine kinase normale mais une ADN polymérase altérée. Ce troisième mécanisme de résistance impliquant l'ADN polymérase du virus de l'herpès simplex est dû à la sélection de mutants codant pour une enzyme altérée, résistante à l'inactivation par l'acyclovir triphosphate.
Le VZV semble manifester une résistance à l'acyclovir via des mécanismes similaires à ceux observés dans le HSV.
Cependant, une enquête clinique limitée n'a révélé aucune preuve d'un changement significatif de la sensibilité in vitro du VZV au traitement par l'acyclovir, bien que des mutants résistants de ce virus puissent être isolés in vitro d'une manière analogue au HSV. L'analyse d'un petit nombre d'isolats cliniques provenant de patients ayant reçu de l'acyclovir par voie orale ou un placebo pour un zona aigu suggère que l'émergence in vivo de VZV résistant peut se produire rarement. Le traitement prolongé par l'acyclovir de patients hautement immunodéprimés atteints d'un syndrome d'immunodéficience acquise et d'un VZV sévère peut entraîner l'apparition de virus résistants.
La résistance croisée à d'autres antiviraux se produit in vitro chez des mutants résistants à l'acyclovir. Les mutants du VHS qui sont résistants à l'acyclovir en raison de l'absence de thymidine kinase virale présentent une résistance croisée à d'autres agents phosphorylés par la thymidine kinase du virus de l'herpès, tels que la bromovinyldésoxyuridine, le ganciclovir et les nucléosides 2'-fluoropyrimidine, tels que le 2'-fluoro -5-iodoarabinosyl-cytosine (FIAC).
La réponse clinique au traitement par l'acyclovir a généralement été bonne pour les patients ayant une immunité normale chez qui le HSV ayant une sensibilité réduite à l'acyclovir a été récupéré, soit avant, pendant ou après le traitement. Cependant, certains groupes de patients, tels que les personnes gravement immunodéprimées (en particulier les receveurs de greffe de moelle osseuse) et ceux qui suivent des régimes suppressifs chroniques, ont été identifiés comme étant le plus souvent associés à l'émergence de souches d'herpès simplex résistantes, qui peuvent ou non accompagner une mauvaise réponse. à la drogue. La possibilité d'apparition de virus moins sensibles doit être reconnue lors du traitement de ces patients, et la surveillance de la sensibilité des isolats cliniques de ces patients doit être encouragée.
En résumé, la relation quantitative entre la sensibilité in vitro du HSV et du VZV à l'acyclovir et la réponse clinique au traitement n'a pas été clairement établie chez l'homme. Des méthodes standardisées de test de sensibilité virale sont nécessaires pour permettre des corrélations plus précises entre la sensibilité virale in vitro et la réponse clinique au traitement par l'acyclovir.
Toxicologie
Études de toxicité aiguë
Souris et rats adultes : La toxicité aiguë de l'acyclovir oral a été déterminée comme indiqué dans le tableau 6.
Rats nouveau-nés, immatures et adultes
Des groupes de 10 rats Charles River CD (Sprague-Dawley) mâles et 10 femelles ont reçu de grandes doses uniques (5 niveaux de dose différents) d'une solution (pH 11,0) d'acyclovir par injection sous-cutanée à 3, 10, 28 et 71 jours. de l'âge. Ils ont été observés pendant 14 jours après le traitement et les valeurs de DL50 ont été calculées par la méthode de Litchfield et Wilcoxon (voir tableau 7 ci-dessous). Cette étude a été réalisée pour déterminer si l'âge au moment de l'exposition affecte la toxicité aiguë de l'acyclovir ; il n'y avait aucune preuve que les jeunes rats étaient plus sensibles que les rats plus âgés aux effets toxiques aigus de l'acyclovir.
Il n'y avait pas de relation apparente entre la durée de survie après traitement et l'âge auquel le traitement a été administré. Les signes cliniques des rats traités à l'âge de 3 et 10 jours comprenaient des cloques cutanées rouges et violettes, des zones bleues, des croûtes, des cicatrices, une peau nécrotique et desquamée, des plaies ouvertes, des tremblements corporels et une alopécie. Chez les rats traités au 28 et 71 jours.
Étude de toxicité orale subchronique
Souris
Quatre groupes constitués chacun de 28 souris Charles River CD-1 (ICR) mâles et 28 femelles ont reçu une dose orale par sonde gastrique pendant 33 jours avec des suspensions d'acyclovir. Les doses quotidiennes étaient de 0, 50, 150 et 450 mg/kg. Des mesures d'hématologie et de chimie clinique ont été effectuées sur 8 souris mâles et 8 souris femelles supplémentaires par groupe (dosées de la même manière) après les première et quatrième semaines d'administration et pendant la 3ème semaine post-dose.
Les concentrations plasmatiques de médicament ont été mesurées dans des échantillons regroupés provenant de 4 souris mâles et 4 femelles supplémentaires par groupe les jours de dose 1, 15 et 30.
Sur la base d'expériences préliminaires avec des rats et des souris, la dose élevée de 450 mg/kg a été choisie pour produire les taux plasmatiques de médicament les plus élevés pouvant être atteints, de manière pratique, par administration orale chez une espèce de rongeur. Les concentrations plasmatiques moyennes du médicament variaient d'environ 3,4 (à la faible dose) à 11,0 (à la dose élevée) μg/mL de plasma une heure après l'administration orale.
Aucun changement dans la santé, le taux de croissance, l'hématologie et les mesures de chimie clinique ne s'est produit qui pourrait être définitivement attribué à l'administration d'acyclovir. Les examens macroscopiques et histopathologiques de 16 rats mâles et 16 rats femelles des groupes à dose élevée et témoin à la fin de la période de dose n'ont rien révélé de remarquable.
Études de toxicité chronique
Étude de toxicité orale à vie chez des rats ayant reçu de l'acyclovir par intubation gastrique
Des rats Charles River CD (Sprague-Dawley) ont reçu des suspensions d'acyclovir par gavage. Il y avait 50 rats mâles et 50 rats femelles à chacune des doses suivantes : 0, 50, 150 et 450 mg/kg. Après 30 et 52 semaines de traitement, 10 rats mâles et 10 rats femelles de chaque groupe ont été autopsiés. Les rats restants ont reçu une dose quotidienne jusqu'à ce que la mortalité naturelle diminue la taille d'un groupe à environ 20 % du nombre d'animaux de ce sexe présents dans les groupes de test au début de l'étude. Tous les rats restants ont été tués et autopsiés lorsque le seuil de 20 % a été atteint. C'était au cours de la semaine 110 pour les rats mâles et de la semaine 122 pour les rats femelles. Les tissus des rats témoins et ceux du groupe à forte dose ont été évalués par microscopie optique. Les tissus des rats des groupes à dose faible et moyenne présentant des masses, des nodules ou des lésions inhabituelles ont également été examinés au microscope optique. Les tissus fixes de rats trouvés morts au cours des 52 premières semaines de l'étude ont également été évalués par microscopie optique.
Aucun signe de toxicose n'a été observé. Des échantillons de plasma ont été prélevés 1,5 heure après l'administration aux jours 7, 90, 209, 369, 771 (hommes uniquement) et 852 (femmes uniquement). Les taux plasmatiques moyens observés chez les mâles recevant la dose élevée (450 mg/kg/jour) aux moments indiqués ci-dessus étaient les suivants : 1,54, 1,63, 1,39, 1,60 et 1,70 μg/mL (6,84, 7,26, 6,17, 7,10 et 7,56 μM) . Les taux plasmatiques moyens correspondants pour les femelles ayant reçu la dose élevée pour les périodes correspondantes étaient de 1,76, 2,38, 2,12, 1,71 et 1,81 μg/mL (7,82, 10,58, 9,44, 7,62 et 8,03 μM). Les concentrations plasmatiques chez les mâles et les femelles à toutes les doses après un an de traitement étaient généralement comparables aux concentrations plasmatiques obtenues lors d'échantillonnages antérieurs. Les valeurs des tests de laboratoire, y compris l'hématologie, la chimie clinique et l'ophtalmoscopie, se situaient toutes dans la plage normale. Il n'y avait pas de lésions macroscopiques ou microscopiques induites par le médicament et il n'y avait aucune preuve que l'acyclovir affectait la survie.
Étude de cancérogénicité orale à vie chez le rat
Il n'y a eu aucun signe de toxicose chez des rats Charles River CD (Sprague-Dawley) (100 rats/sexe/groupe de dose) ayant reçu de l'acyclovir par gavage oral à 50, 150 et 450 mg/kg dans une étude de cancérogénicité orale à vie. Les concentrations plasmatiques moyennes obtenues chez les mâles recevant la dose élevée 1,5 heure après l'administration à divers moments d'échantillonnage au cours de l'étude étaient les suivantes : 1,54, 1,63, 1,39, 1,60 et 1,70 μg/mL (6,84, 7,26, 6,17, 7,10 et 7,56 μM) aux jours 7, 90, 209, 369 et 771, respectivement. Les valeurs moyennes correspondantes pour les femelles ayant reçu la dose élevée étaient de 1,76, 2,38, 2,12, 1,71 et 1,81 μg/mL (7,82, 10,58, 9,44, 7,62 et 8,03 μM) aux jours 7, 90, 209, 369 et 852, respectivement.
Les valeurs des tests de laboratoire clinique, y compris l'hématologie, la chimie clinique, l'analyse d'urine, le poids corporel, la consommation alimentaire et l'ophtalmoscopie, étaient toutes dans les limites normales. Il n'y avait pas de lésions macroscopiques ou microscopiques induites par le médicament et il n'y avait aucune preuve que l'acyclovir affectait la survie, les tendances temporelles de l'incidence des tumeurs ou le nombre de tumeurs bénignes ou malignes.
La plupart des rats relativement peu nombreux trouvés morts ou moribonds au cours des 52 premières semaines de cette étude ont subi des accidents de dosage, comme en témoignent les découvertes post-mortem de perforation œsophagienne provoquant un épanchement pleural, une pneumonie ou une médiastinite.
Étude de cancérogénicité orale à vie chez la souris
Il n'y a eu aucun signe de toxicose chez les souris Charles River CD-1 (ICR) (115 souris/sexe/groupe de dose) ayant reçu de l'acyclovir par gavage oral à 50, 150 et 450 mg/kg/jour dans une étude de cancérogénicité orale à vie. Les concentrations plasmatiques moyennes obtenues chez les mâles recevant la dose élevée 1,5 heure après l'administration à divers moments d'échantillonnage au cours de l'étude étaient les suivantes : 2,83, 3,17 et 1,82 μg/mL (12,59, 14,10 et 8,10 μM) aux jours 90, 365 et 541, respectivement. Les valeurs moyennes correspondantes pour les femelles ayant reçu la dose élevée étaient de 9,81, 5,85 et 4,0 μg/mL (43,60, 26,0 et 17,79 μM).
Les valeurs des tests de laboratoire clinique, y compris l'hématologie, le poids corporel et la consommation alimentaire, se situaient toutes dans les limites normales. Il n'y avait pas de lésions macroscopiques ou microscopiques induites par le médicament. Les souris femelles ayant reçu 150 et 450 mg/kg d'acyclovir ont survécu significativement plus longtemps que les souris femelles témoins ; la survie des mâles traités était comparable à la survie des mâles témoins. Les schémas d'incidence des tumeurs et le nombre de tumeurs pour les néoplasmes bénins ou malins n'ont pas été affectés par le traitement par l'acyclovir.
Étude de toxicité orale chronique de 12 mois chez le chien
Des chiens Beagle de race pure ont reçu 0, 15, 45 ou 150 mg/kg/jour d'acyclovir chaque jour pendant les deux premières semaines d'une étude d'un an. Il y avait 9 mâles et 9 femelles dans chaque groupe de test. Les chiens ont reçu des capsules de gélatine contenant la dose appropriée. Ils ont été traités tid, donc les doses administrées à chacune des trois périodes de dose équidistantes étaient de 0, 5, 15 et 50 mg/kg. Les doses de 45 et 150 mg/kg ont provoqué des diarrhées, des vomissements, une diminution de la consommation alimentaire et une perte de poids chez les chiens mâles et femelles au cours des deux premières semaines de l'étude. Pour cette raison, au cours de la troisième semaine de l'étude, la décision a été prise de diminuer les doses moyenne et élevée à 30 et 60 mg/kg/jour (10 et 20 mg/kg tid). La faible dose de 15 mg/kg/jour (5 mg/kg tid) était inchangée. Les chiens ayant reçu 60 mg/kg/jour vomissaient occasionnellement et souffraient occasionnellement de diarrhée, mais s'en sortaient bien pendant toute la durée du test, et les valeurs de gain de poids corporel et de consommation alimentaire étaient comparables aux valeurs témoins.
Au cours de la toxicose induite par les fortes doses d'acyclovir, les taux plasmatiques du médicament étaient probablement très élevés (comme l'indiquent les valeurs moyennes initiales de 24,0 μg/mL (106,6 μM) pour les hommes recevant la dose élevée et de 17,4 μg/mL (77,2 μM) pour les femelles recevant la dose élevée lorsqu'elle est déterminée 1 heure après la troisième dose le jour 1 de l'étude). Lorsqu'ils ont été mesurés au jour 15, les taux plasmatiques d'acyclovir chez les chiens recevant une dose élevée (150 mg/kg/jour) étaient encore très élevés, mais ils ont diminué plus tard lorsque les doses ont été réduites. Les valeurs des concentrations plasmatiques après 12 mois de traitement étaient généralement comparables aux valeurs enregistrées après 1, 3 et 6 mois de traitement. Ainsi, il n'y avait aucune indication d'un métabolisme accru de l'acyclovir à la suite d'un traitement chronique.
Au cours de la 13e semaine, certains chiens mâles et femelles aux doses moyenne et élevée ont présenté les signes suivants : sensibilité des pattes avant, érosion des coussinets plantaires, cassure et relâchement des ongles. La régénération des ongles perdus a commencé quelques semaines plus tard. Les ongles régénérés au bout de 6 mois (lorsque 3 mâles et 3 femelles de chaque groupe ont été tués pour un sacrifice intermédiaire) et à la fin de l'étude étaient généralement de bonne qualité. Il n'y a jamais eu de signes d'effet sur les pattes ou les ongles chez les chiens du groupe à faible dose (15 mg/kg/jour).
Il est admis qu'une lésion de l'épithélium corial qui produit la kératine de l'ongle peut entraîner l'arrêt de la production de kératine et la production de kératine anormale. La toxicose transitoire induite par les fortes doses (45 et 150 mg/kg/jour) d'acyclovir administrées pendant les deux premières semaines de l'étude peut avoir affecté l'épithélium corial. S'il y avait un effet transitoire sur l'épithélium corial (éventuellement lié à des effets directs ou secondaire à une maladie induite par le médicament au cours des deux premières semaines de l'étude), la perte ultérieure de l'ongle pourrait être une séquelle. Aucun effet perceptible sur d'autres tissus producteurs de kératine ou contenant de la kératine n'a été observé. Il faut souligner que les altérations des ongles semblaient liées à la toxicose transitoire induite par les doses de 50 et 150 mg/kg/jour testées pendant les deux premières semaines de l'étude et non aux doses de 30 et 60 mg/kg niveaux de dose / jour testés par la suite.
Il n'y a eu aucune modification importante induite par le médicament dans les valeurs des tests biochimiques sériques, des analyses d'urine et des tests électrocardiographiques effectués à des intervalles appropriés au cours de cette étude. Les valeurs d'albumine sérique et de protéines totales ont légèrement diminué chez les chiens traités à 30 et 60 mg/kg/jour pendant 6 et 12 mois. Cependant, toutes les valeurs de ces paramètres sont restées dans les limites acceptées comme normales.
À l'exception des altérations résiduelles de la vieille kératine au bout des griffes, il n'y avait aucun signe d'effets liés au traitement dans aucun des tissus examinés au microscope optique. Il n'y avait pas non plus de modifications significatives des valeurs des organes pesés à l'autopsie. Ainsi, des doses allant jusqu'à 60 mg/kg/jour ont été bien tolérées pendant un an. Le niveau de dose « sans effet sur la dose » d'acyclovir était de 15 mg/kg/jour (5 mg/kg tid) ; cependant, les seuls effets indésirables à 30 ou 60 mg/kg/jour étaient des modifications des ongles et des coussinets plantaires (30 et 60 mg/kg/jour) et des signes gastro-intestinaux légers (60 mg/kg/jour).
Études de reproduction
Tératologie – Rats
L'acyclovir a été administré à des rats femelles gravides ARS Sprague-Dawley par injection sous-cutanée pendant la période d'organogenèse (du jour 6 au jour 15 de la gestation) à des doses de 0,0, 6,0, 12,5 et 25,0 mg/kg de poids corporel deux fois par jour.
Les critères évalués pour l'effet composé comprenaient le poids corporel maternel, les gains de poids, l'apparence et le comportement, les taux de survie, les changements oculaires, les taux de grossesse et les données sur la reproduction. La viabilité et le développement de la progéniture ont également été évalués.
En plus des mesures ci-dessus, des animaux désignés ont été sacrifiés 1 heure après la première dose au jour 15 afin de prélever des échantillons de sang maternel, de liquide amniotique et de fœtus pour des mesures de concentration de médicament. Les valeurs moyennes de ces échantillons sont répertoriées dans le tableau 8.
Les valeurs obtenues pour le plasma représenteraient environ 30 % des taux plasmatiques initiaux à en juger par la demi-vie plasmatique chez les rongeurs.
Aucun effet attribuable à l'administration d'acyclovir n'a été noté dans les comparaisons des valeurs de poids corporel maternel, de l'apparence et du comportement, des taux de survie, des taux de grossesse ou des efficacités d'implantation. De plus, aucune différence liée au composé n'a été notée dans les évaluations de la taille, du sexe et du développement du fœtus.
Bien que les incidences de résorption et de viabilité fœtale se situent dans la plage de variabilité normale dans tous les groupes, des incidences légèrement plus élevées de résorptions ont été notées chez les animaux à forte dose sacrifiés aux jours 15 et 19 de la gestation ; cependant, aucune tendance claire liée à la dose ne s'est manifestée.
Par conséquent, l'acyclovir n'a pas été considéré comme tératogène ou embryotoxique lorsqu'il a été administré à des rats à des doses allant jusqu'à 50,0 mg/kg de poids corporel par jour pendant l'organogenèse.
Tératologie – Lapins
Une étude de tératologie a été réalisée chez des lapins blancs de Nouvelle-Zélande en utilisant essentiellement le même schéma expérimental que chez le rat, sauf que le dosage était du jour 6 au jour 18 de la gestation. De plus, la collecte de fœtus, de liquide amniotique et d'échantillons de sang maternel a eu lieu le jour 18 plutôt que le jour 15.
Aucun signe de toxicité maternelle n'a été observé à aucune dose, mais il y avait une efficacité d'implantation inférieure statistiquement significative (p
Des concentrations d'acyclovir ont été détectées dans des échantillons de plasma et de liquide amniotique, ainsi que dans des homogénats de tissus fœtaux. Tous les échantillons ont été prélevés une heure après la première dose au jour 18 de la gestation. Les concentrations de médicament dans le liquide amniotique étaient considérablement plus élevées que celles du plasma (voir tableau 9).
Reproduction – Fertilité
L'acyclovir s'est avéré ne pas altérer la fertilité ou la reproduction dans des groupes de 15 souris mâles et 30 souris femelles dans une étude de fertilité sur deux générations. Les souris de cette étude ont reçu de l'acyclovir par intubation gastrique à des doses de 50, 150 et 450 mg/kg/jour. Les mâles ont reçu une dose pendant 64 jours consécutifs avant l'accouplement et les femelles pendant 21 jours avant l'accouplement.
Dans une étude de fertilité chez le rat où des groupes de 20 rats mâles et 20 rats femelles ont reçu 0, 12,5, 25,0 et 50,0 mg/kg/jour par injection sous-cutanée, l'acyclovir n'a pas eu d'effet sur l'accouplement ou la fertilité. Les mâles ont reçu une dose pendant 60 jours avant l'accouplement et jusqu'à ce que leur calendrier d'accouplement soit terminé. Les rats femelles ont reçu une dose pendant 14 jours avant l'accouplement et jusqu'au jour 7 de la grossesse. À 50 mg/kg/jour sc, il y a eu une augmentation statistiquement significative de la perte post-implantation, mais aucune diminution concomitante de la taille de la portée.
Chez 25 lapines traitées par voie sous-cutanée avec 50 mg/kg/jour d'acyclovir les jours 6 à 18 de la gestation, il y a eu une diminution statistiquement significative de l'efficacité d'implantation mais aucune diminution concomitante de la taille de la portée. Il y avait également une augmentation liée à la dose du nombre de fœtus avec des côtes surnuméraires dans tous les groupes traités par le médicament. Cette augmentation n'était pas liée à la dose lorsque l'incidence des côtes surnuméraires par portée a été examinée.
Chez 15 lapines traitées par voie intraveineuse avec 50 mg/kg/jour d'acyclovir les jours 6 à 18 de la gestation, il n'y a eu aucun effet sur l'efficacité d'implantation ou la taille de la portée.
Dans une étude périnatale et postnatale chez le rat (20 rats femelles par groupe), l'acyclovir a été administré par voie sous-cutanée à raison de 0, 12,5, 25 et 50 mg/kg/jour du 17e jour de gestation au 21e jour postpartum. À 50 mg/kg/jour sc, il y a eu une diminution statistiquement significative du nombre moyen de corps jaunes, de sites d'implantation totaux et de fœtus vivants dans la génération F1. Bien que non statistiquement significative, il y a également eu une diminution liée à la dose du nombre moyen de fœtus vivants et de sites d'implantation à 12,5 mg/kg/jour et 25 mg/kg/jour sc
Dans une étude de dosage portant sur 5 lapines, l'administration intraveineuse d'acyclovir à la dose de 100 mg/kg/jour du 6e au 8e jour de gestation, dose connue pour provoquer une néphropathie obstructive, a provoqué une augmentation significative des résorptions fœtales et une diminution correspondante de la taille de la portée. À une dose intraveineuse maximale tolérée de 50 mg/kg/jour chez le lapin, aucun effet sur la reproduction lié au médicament n'a été observé.
Dans une étude de toxicité subchronique où des groupes de 20 rats mâles et 20 rats femelles ont reçu des doses intrapéritonéales d'acyclovir à 0, 20, 80 ou 320 mg/kg/jour pendant un mois, et suivis pendant une période post-dose d'un mois, il y avait atrophie. Certaines preuves histologiques de récupération de la production de spermatozoïdes étaient évidentes 30 jours après la dose, mais ce n'était pas suffisamment de temps pour démontrer une réversibilité complète.
Des groupes de 25 rats mâles et 25 rats femelles ont reçu des doses intrapéritonéales d'acyclovir à 0, 5, 20 ou 80 mg/kg/jour pendant 6 mois. Dix rats mâles et 10 rats femelles dans chaque groupe ont été poursuivis sans dose pendant 13 semaines. L'atrophie testiculaire était limitée aux rats ayant reçu une dose élevée de 80 mg/kg/jour pendant 6 mois. Les données sur le poids des organes et la microscopie optique ont défini la réversibilité complète de l'atrophie testiculaire à la fin de la période de récupération post-dose.
Dans une étude de 31 jours chez le chien (16 mâles et 16 femelles par groupe) où l'acyclovir a été administré par voie intraveineuse à des niveaux de 50, 100 et 200 mg/kg/jour, les testicules étaient normaux chez les chiens à 50 mg/kg. Des doses de 100 ou 200 mg/kg/jour ont causé la mort de certains chiens en raison d'effets cytostatiques (moelle osseuse et épithélium gastro-intestinal) et de testicules aspermiques ou de testicules avec des tubules aspermiques dispersés. On ne peut pas exclure que le changement testiculaire ait pu être primaire, cependant, des changements similaires peuvent être observés à la suite d'un stress sévère chez les chiens moribonds.
Études de toxicité pour le développement
Rats nouveau-nés - Étude subchronique
De l'acyclovir dissous dans une solution saline stérile à 0,4 % a été administré par injection sous-cutanée à des rats nouveau-nés Charles River CD (Sprague-Dawley) pendant 19 jours consécutifs, en commençant le 3e jour post-partum. Les doses testées étaient de 0, 5, 20 et 80 mg/kg de poids corporel. Il y avait 12 portées (chacune consistant en 5 mâles et 5 femelles nouveau-nés allaitant la mère naturelle) à chaque niveau de dose. Les barrages n'ont pas été traités. Les nouveau-nés ont été retirés de chaque groupe pour une autopsie et une évaluation microscopique d'une grande variété de tissus, y compris les yeux et plusieurs sections du cerveau, après avoir été traités pendant 5, 12 ou 19 jours et après une période sans médicament de 3 semaines après la dose ( à ce moment-là, ils étaient âgés de 45 jours). Des tests hématologiques (hémoglobine, hématocrite, numération érythrocytaire, leucocytaire et différentielle) et de chimie clinique (BUN) ont été effectués après 16 jours de traitement et répétés 18 jours après l'administration de la dernière (19e) dose.
Du sang a été prélevé sur certains nouveau-nés 30 minutes après le traitement au jour 1, au jour 9 et à la fin de la période de dose pour la détermination des concentrations d'acyclovir dans le plasma. La plus grande concentration d'acyclovir dans le plasma était de 99,1 μg/mL (440,5 μM) trouvée dans le plasma regroupé prélevé chez 6 nouveau-nés femelles à forte dose (80 mg/kg) 30 minutes après l'administration de la première dose. Le traitement par l'acyclovir n'a pas augmenté la mortalité en période néonatale.
Les rats du groupe à faible dose ont pris autant de poids corporel que les rats témoins respectifs. Des réductions significatives (p
Les examens des yeux et la microscopie optique n'ont pas révélé d'effets indésirables sur le développement oculaire. Il convient de souligner qu'il n'y avait aucune preuve morphologique ou fonctionnelle d'effets indésirables sur le développement du cerveau ou d'autres parties du système nerveux central. Ainsi, l'acyclovir est nettement différent de la cytosine arabinoside qui a été signalée comme produisant une dysplasie cérébelleuse et rétinienne importante chez les rats nouveau-nés.
Mutagénicité et autres études à court terme
L'acyclovir a été testé pour son potentiel mutagène dans un certain nombre de systèmes in vitro et in vivo : 10 novembre 2014 Page 27 de 38
microbien
L'acyclovir a été testé pour l'activité mutagène dans le test sur plaque d'Ames Salmonella ; dans une modification de préincubation du test d'Ames ; dans le test de réparation de l'ADN polA+/polA- de Rosenkrantz E. coli ; et chez l'eucaryote S. cerevisiae, D-4. Toutes les études ont été réalisées à la fois en présence et en l'absence d'activation métabolique exogène chez les mammifères. L'acyclovir n'a donné aucune réponse positive dans aucun de ces systèmes.
Les études précédentes sur Salmonella ont été étendues à des concentrations extrêmement élevées afin d'obtenir une toxicité. Aucun effet positif n'a été observé en présence ou en l'absence d'activation métabolique exogène chez les mammifères, à des concentrations d'acyclovir allant jusqu'à 300 mg/plaque ou 80 mg/mL.
Systèmes mammifères
L'acyclovir a été testé pour l'activité mutagène dans des cellules de lymphome de souris L5178Y en culture, hétérozygotes au locus de la thymidine kinase (TK), en mesurant le taux de mutation vers le déficit en TK (TK+/- → TK-/- ; des études supplémentaires ont été réalisées au HGPRT locus et au marqueur de résistance à l'ouabaïne dans ces mêmes cellules Toutes les études ont été réalisées en présence et en l'absence d'activation métabolique exogène chez les mammifères Le composé testé était mutagène au locus TK à des concentrations élevées (400 -2 400 μg/mL) (En comparaison, la limite supérieure des concentrations plasmatiques maximales d'acyclovir après une administration orale de 200 mg q4h est de 0,9 μg/mL. Elle était négative au locus HGPRT et au marqueur de résistance à l'ouabaïne. Des résultats identiques ont été obtenus avec et sans activation métabolique.
Des résultats non concluants sans réponse apparente liée à la dose ont été obtenus lorsque la mutagénicité de l'acyclovir a été étudiée à chacun des 3 loci (APRT, HGPRT et résistance à l'ouabaïne) dans des cellules ovariennes de hamster chinois (CHO), à la fois en présence et en l'absence d'activation métabolique exogène.
Il a été démontré que l'acyclovir, à une concentration de 50 μg/mL (222 μM) pour une exposition de 72 heures, provoque une augmentation statistiquement significative de l'incidence des foyers morphologiquement transformés résultant du traitement des cellules BALB/C-3T3 in vitro chez l'absence d'activation métabolique exogène. Il a été démontré que les foyers morphologiquement transformés se développent sous forme de tumeurs après transplantation chez des souris sevrées syngéniques immunosupprimées. Les tissus tumoraux ont été diagnostiqués comme étant soit des sarcomes indifférenciés, soit des lymphosarcomes.
L'acyclovir, à des concentrations comprises entre 8 et 64 μg/mL pendant 18 heures d'exposition, n'a induit aucun foyer morphologiquement transformé parmi les cellules C3H/10T ½ traitées in vitro en l'absence d'activation métabolique exogène.
L'acyclovir, à des concentrations de 62,5 et 125 μg/mL pour une exposition de 48 heures, n'a induit aucune aberration chromosomique dans les lymphocytes humains en culture en l'absence d'activation métabolique exogène. À des concentrations plus élevées, 250 et 500 μg/mL pour une exposition de 48 heures, l'acyclovir a provoqué une augmentation significative de l'incidence des cassures chromosomiques. Il y avait aussi une diminution significative liée à la dose de l'indice mitotique avec l'exposition à l'acyclovir.
L'acyclovir, à des doses de 25 et 50 mg/kg/jour ip pendant 5 jours consécutifs, n'a pas produit d'effet létal dominant chez les souris mâles BKA (CPLP). De plus, il n'y avait aucune preuve d'un effet létal dominant sur les souris mâles et femelles Charles River CD-1 (ICR) traitées par voie orale à des doses de 50, 150 et 450 mg/kg/jour comme résumé pour l'étude de reproduction/fertilité sur deux générations. .
L'acyclovir, à des doses intrapéritonéales uniques de 25, 50 et 100 mg/kg, n'a pas provoqué d'aberrations chromosomiques dans les cellules de la moelle osseuse de hamsters chinois lorsqu'il a été examiné 24 heures après l'administration. A des doses néphrotoxiques plus élevées (500 et 1 000 mg/kg), un effet blastogène a été observé. (Une dose intrapéritonéale de 500 mg/kg produit des concentrations plasmatiques maximales moyennes chez les hamsters chinois de 611 μg/mL (2,72 mM), ce qui est 680 fois plus élevé que la limite supérieure des concentrations plasmatiques maximales humaines lors d'une administration orale de 200 mg q4h).
L'acyclovir, à des doses intraveineuses uniques de 25, 50 et 100 mg/kg, n'a pas provoqué d'aberrations chromosomiques dans les cellules de la moelle osseuse des rats mâles et femelles lorsqu'il a été examiné 6, 24 et 48 heures après le traitement.
Ainsi, toutes ces études ont montré que l'acyclovir ne provoque pas de mutations monogéniques mais est capable de casser des chromosomes.
Études d'immunotoxicologie
L'acyclovir a été soumis à un certain nombre de tests immunologiques in vitro et in vivo.
Dans deux tests in vivo, la cytotoxicité médiée par les lymphocytes et la chimiotaxie des neutrophiles, l'acyclovir n'a montré aucun effet inhibiteur à des concentrations aussi élevées que 135 μg/mL (600 μM). Le composé a inhibé la formation de rosettes d'environ 50 % à 0,9 μg/mL (4 μM).
Dans quatre tests in vivo chez la souris qui ont mesuré l'immunité à médiation cellulaire (cytotoxicité cellulaire dépendante du complément, cytotoxicité cellulaire indépendante du complément, hypersensibilité retardée et réaction du greffon contre l'hôte), l'acyclovir n'a montré aucun effet inhibiteur à des doses uniques allant jusqu'à 200 mg/kg administrées au jour 2 après la stimulation antigénique.
Quatre doses quotidiennes de 100 mg/kg/jour n'ont eu aucun effet significatif sur les plaques d'hémolysine de Jerne ou sur les anticorps circulants au jour 7 après la stimulation antigénique. Lorsque les plaques d'hémolysine de Jerne et les titres d'anticorps ont été examinés quatre jours après la provocation antigénique et un jour après la dernière dose de médicament, 100 mg/kg n'ont montré qu'un léger effet suppresseur. Cependant, 200 mg/kg ont produit une certaine perte de poids (-2,2 g), une réduction modérée du nombre de plaques d'hémolysine de Jerne (PFC/rate ont été réduits à 33 % du contrôle, PFC/107 WBC à 46,5 % du contrôle). Cependant, il n'y avait qu'une faible réduction du titre d'hémagglutinine circulante (de 8,3 à 6,5) et du titre d'hémolysine circulante (de 9,5 à 8,3) à 200 mg/kg.
Dans des expériences sur des souris conçues pour tester si l'acyclovir potentialiserait l'effet immunosuppresseur de l'azathioprine sur la formation d'anticorps, il a été constaté que les effets des deux médicaments n'étaient qu'additifs. Seule la dose de 200 mg/kg d'acyclovir a montré une suppression accrue de la réponse anticorps lorsqu'elle était administrée en association avec l'azathioprine à des doses supérieures à 25 mg/kg.
Des études ont été menées pour évaluer l'influence de l'acyclovir in vitro sur la fonction des lymphocytes humains. Les effets inhibiteurs sur la blastogenèse n'ont été observés que dans les tests examinant les concentrations maximales de mitogènes puissants, la phytohémagglutinine (PHA) et la concanavaline A (Con A), et uniquement à des concentrations de médicament supérieures à 50 μg/mL (222 μM) et étaient bien moindres avec monilia et les antigènes de l'anatoxine tétanique, où la réponse blastogène est généralement moins vigoureuse. Il y avait très peu d'effet sur la cytotoxicité ou la production de LIF sauf à des concentrations de 200 μg/mL (890 μM) où il a déjà été démontré qu'il y a un effet cytotoxique direct. Ces concentrations inhibitrices sont bien supérieures aux niveaux anticipés des doses sélectionnées pour l'application clinique et plus de 1 000 fois supérieures à la concentration requise pour inhiber la multiplication de l'herpèsvirus in vitro.
L'effet de l'acyclovir sur les cellules humaines a été mesuré. Une concentration de 11,2 à 22,5 μg/mL (50-100 μM) inhibe la division des fibroblastes dans une mesure variable, selon la conception expérimentale et la confluence de la monocouche. L'ampleur de cet effet était inférieure à celle causée par l'adénine arabinoside ou l'interféron leucocytaire humain lorsque ces trois agents antiviraux ont été comparés à des concentrations cliniquement pertinentes. L'acyclovir a également inhibé l'incorporation de la thymidine par les cellules mononucléaires du sang périphérique stimulées par la PHA ou trois antigènes différents du virus de l'herpès. Une courbe dose-réponse linéaire a été observée avec ces cellules, et leur prolifération a été inhibée à 50 % par 22,5 μg/mL (100 μM) d'acyclovir. L'inhibition a été exercée sur la prolifération des lymphocytes T sans effet apparent sur la libération de lymphokines ou sur la fonction des monocytes.
Il convient également de mentionner qu'il n'y avait aucune preuve d'effets indésirables sur le système immunitaire dans les tests détaillés subchroniques et chroniques sur les animaux couverts plus tôt dans ce résumé, sauf à des doses excessivement élevées (50 à 100 mg/kg bid) chez les chiens où une hypoplasie lymphoïde marquée eu lieu.
RÉFÉRENCES
1. Balfour HH, Jr., Kelly JM, Suarez CS, Heussner RC, Englund JA, Crane DD et al. Traitement de la varicelle par l'acyclovir chez des enfants par ailleurs en bonne santé. J Pediatr 1990; 116(4):633-639.
2. Balfour HH, Jr., Rotbart HA, Feldman S, Dunkle LM, Feder HM, Jr., Prober CG et al. Traitement de la varicelle par l'acyclovir chez des adolescents par ailleurs en bonne santé. Le groupe d'étude collaborative sur l'acyclovir et la varicelle. J Pediatr 1992; 120(4 Pt 1):627-633.
3. Barry DW, Blum MR. Antiviraux : acyclovir, dans Recent Advances in Clinical Pharmacology. Turner P, Shand DG (eds) Churchill Livingstone, Édimbourg 1983.
4. Barry DW, Nusinoff-Lehrman S. Résistance virale dans la pratique clinique : résumé de cinq années d'expérience avec l'acyclovir. Approches pharmacologiques et cliniques des virus de l'herpès et de la chimiothérapie virale, Aiso, Japon, 10-13 septembre 1984.
5. Barry DW, Nusinoff-Lehrman S, Ellis MN, Biron KK, Furman PA. Résistance virale, expérience clinique. Scand J Infect Dis Suppl 1985; 47:155-164.
6. Barry DW, Nusinoff-Lehrman S. Résistance virale dans la pratique clinique : résumé de cinq années d'expérience avec l'acyclovir. Actes du Symposium international sur les approches pharmacologiques et cliniques des virus de l'herpès et de la chimiothérapie virale, Elsever, Amsterdam 1985;269-270.
7. Biron KK, Elion GB. Effet de l'acyclovir combiné avec d'autres agents antiherpétiques sur le virus varicelle-zona in vitro. Am J Med 1982; 73(1A):54-57.
8. Boelaert J, Schurgers M, Daneels R, Van Landuyt HW, Weatherley BC. Pharmacocinétique à doses multiples d'acyclovir intraveineux chez des patients sous dialyse péritonéale continue ambulatoire. J Antimicrob Chemother 1987; 20(1):69-76.
9. Bryson YJ, Dillon M, Lovett M, Acuna G, Taylor S, Cherry JD et al. Traitement des premiers épisodes d'infection génitale par le virus de l'herpès simplex par l'acyclovir par voie orale. Un essai contrôlé randomisé en double aveugle chez des sujets normaux. N Engl J Med 1983; 308(16):916-921.
10. Burns WH, Saral R, Santos GW, Laskin OL, Lietman PS, McLaren C et al. Isolement et caractérisation du virus Herpes simplex résistant après traitement à l'acyclovir. Lancette 1982 ; 1(8269):421-423.
11. Christophers J, Sutton RN. Caractérisation d'isolats cliniques de virus de l'herpès simplex résistants et sensibles à l'acyclovir provenant d'un patient immunodéprimé. J Antimicrob Chemother 1987; 20(3):389-398.
12. Cole NL, Balfour HH, Jr. Le virus varicelle-zona ne devient pas plus résistant à l'acyclovir pendant le traitement. J Infect Dis 1986; 153(3):605-608.
13. Collins P, DJ Bauer. L'activité in vitro contre le virus de l'herpès de la 9-(2-hydroxyéthoxyméthyl)guanine (acycloguanosine), un nouvel agent antiviral. J Antimicrob Chemother 1979; 5(4):431-436.
14. Collins P, Oliver NM. Surveillance de la sensibilité des isolats du virus de l'herpès simplex provenant de patients recevant de l'acyclovir. J Antimicrob Chemother 1986; 18 Supplément B:103-112.
15. Collins P. Sensibilité virale suite à l'introduction de l'acyclovir. Am J Med 1988; 85(2A):129-134.
16. Collins P, Larder BA, Oliver NM, Kemp S, Smith IW, Darby G. Caractérisation d'un mutant de l'ADN polymérase du virus de l'herpès simplex d'un patient gravement immunodéprimé recevant de l'acyclovir. J Gen Virol 1989; 70 (Partie 2):375-382.
17. Crumpacker CS, Schnipper LE, Zaia JA, Levin MJ. Inhibition de la croissance par l'acycloguanosine des virus de l'herpès isolés d'infections humaines. Agents antimicrobiens Chemother 1979 ; 15(5):642-645.
18. Crumpacker CS, Schnipper LE, Marlowe SI, Kowalsky PN, Hershey BJ, Levin MJ. Résistance aux médicaments antiviraux du virus de l'herpès simplex isolé chez un patient traité par l'acyclovir. N Engl J Med 1982; 306(6):343-346.
19. Darby G, Inglis MM, Larder BA. Mécanismes de résistance aux analogues nucléosidiques inhibiteurs du virus de l'herpès simplex. 6th Int Congr Virol 1984;(Résumé #W34-5).
20. De Clercq E, Descamps J, Verhelst G, Walker RT, Jones AS, Torrence PF et al. Efficacité comparative des médicaments anti-herpès contre différentes souches du virus de l'herpès simplex. J Infect Dis 1980; 141(5):563-574.
21. De Clercq E. Efficacité comparative des médicaments anti-herpès dans différentes lignées cellulaires. Agents antimicrobiens Chemother 1982 ; 21(4):661-663.
22. Dekker C, Ellis MN, McLaren C, Hunter G, Rogers J, Barry DW. La résistance aux virus en pratique clinique. J Antimicrob Chemother 1983; 12 Supplément B:137-152.
23. Douglas JM, Davis LG, Remington ML, Paulsen CA, Perrin EB, Goodman P et al. Un essai en double aveugle contrôlé par placebo sur l'effet de l'acyclovir oral administré de manière chronique sur la production de sperme chez les hommes atteints d'herpès génital fréquemment récurrent. J Infect Dis 1988 mars ; 157:588-93.
24. Douglas JM, Critchlow C, Benedetti J, Mertz GJ, Connor JD, Hintz MA et al. Une étude en double aveugle de l'acyclovir par voie orale pour la suppression des récidives de l'infection par le virus de l'herpès simplex génital. N Engl J Med 1984; 310(24):1551-1556.
25. Dunkle LM, Arvin AM, Whitley RJ, Rotbart HA, Feder HM, Jr., Feldman S et al. Un essai contrôlé de l'acyclovir pour la varicelle chez les enfants normaux. N Engl J Med 1991; 325(22):1539-1544.
26. Ellis MN, Keller PM, Martin JL, Strauss SE, Nusinoff-Lehrman S et al. Caractérisation d'un isolat clinique HSV-2 contenant un mutant résistant à l'ACV qui produit une thymidine kinase avec une spécificité de substrat altérée. Ninth Int Herpesvirus Workshop, Seattle, Washington, 24-29 août 1984.
27. Ellis MN, Keller PM, Fyfe JA, Martin JL, Rooney JF, Straus SE et al. Isolat clinique du virus de l'herpès simplex de type 2 qui induit une thymidine kinase avec une spécificité de substrat altérée. Agents antimicrobiens Chemother 1987 ; 31(7):1117-1125.
28. Englund JA, Zimmerman ME, Swierkosz EM, Goodman JL, Scholl DR, Balfour HH, Jr. Virus de l'herpès simplex résistant à l'acyclovir. Une étude dans un centre de soins tertiaires. Ann Intern Med 1990 ; 112(6):416-422.
29. Erlich KS, Jacobson MA, Koehler JE, Follansbee SE, Drennan DP, Gooze L et al. Thérapie Foscarnet pour les infections graves à virus herpès simplex de type 2 résistant à l'acyclovir chez les patients atteints du syndrome d'immunodéficience acquise (SIDA). Un procès incontrôlé. Ann Intern Med 1989 ; 110(9):710-713.
30. Erlich KS, Mills J, Chatis P, Mertz GJ, Busch DF, Follansbee SE et al. Infections par le virus de l'herpès simplex résistant à l'acyclovir chez les patients atteints du syndrome d'immunodéficience acquise. N Engl J Med 1989; 320(5):293-296.
31. Champ HJ, Darby G, Wildy P. Isolement et caractérisation des mutants résistants à l'acyclovir du virus de l'herpès simplex. J Gen Virol 1980; 49(1):115-124.
32. Champ HJ. Le problème de la résistance induite par les médicaments chez les virus, dans Problems of Antiviral Therapy. Stuart-Harris CH, Oxford J (Eds) Academic Press, Londres 1983.
33. Fyfe K. Schémas de récurrence de l'herpès génital après l'arrêt de plus de 5 ans de suppression chronique de l'acyclovir. VIII Int Conf AIDS/III Std Wrld Cong 1992 ; (B240).
34. Huff JC, Bean B, Balfour HH, Jr., Laskin OL, Connor JD, Corey L et al. Thérapie du zona avec de l'acyclovir par voie orale. Am J Med 1988; 85(2A):84-89.
35. Jacobson MA, Berger TG, Fikrig S, Becherer P, Moohr JW, Stanat SC et al. Infection par le virus varicelle-zona résistante à l'acyclovir après un traitement chronique par acyclovir par voie orale chez des patients atteints du syndrome d'immunodéficience acquise (SIDA). Ann Intern Med 1990 ; 112(3):187-191.
36. Kaplowitz LG, Baker D, Gelb L, Blythe J, Hale R, Frost P et al. Traitement continu prolongé à l'acyclovir d'adultes normaux atteints d'une infection par le virus de l'herpès simplex génital fréquemment récurrente. Le groupe d'étude sur l'acyclovir. JAMA 1991; 265(6):747-751.
37. Krasny HC, Liao SH, de Miranda P, Laskin OL, Whelton A, Lietman PS. Influence de l'hémodialyse sur la pharmacocinétique de l'acyclovir chez les patients insuffisants rénaux chroniques. Am J Med 1982; 73(1A):202-204.
38. Kurtz T. Innocuité et efficacité du traitement suppressif à long terme au cyclovir de l'herpès génital fréquemment récurrent : résultats de l'année 5. 30th Intersci Conf Antimicrob Agents Chemother 1990;270.
39. Laskin OL, Longstreth JA, Whelton A, Krasny HC, Keeney RE, Rocco L et al. Effet de l'insuffisance rénale sur la pharmacocinétique de l'acyclovir. Am J Med 1982; 73(1A):197-201.
40. Lau RJ, Emery MG, Galinsky RE. Accumulation inattendue d'acyclovir dans le lait maternel avec estimation de l'exposition du nourrisson. Obstet Gynecol 1987; 69(3 Pt 2):468-471.
41. Lehrman SN, Douglas JM, Corey L, Barry DW. Herpès génital récurrent et thérapie orale suppressive à l'acyclovir. Relation entre les résultats cliniques et la sensibilité aux médicaments in vitro. Ann Intern Med 1986 ; 104(6):786-790.
42. Marlowe S, Douglas J, Corey L, Schnipper L, Crumpacker C. Sensibilité des isolats génitaux du VHS après acyclovir oral. 24th Interscience Conf Antimicrob Ag Chemother, Washington, DC, 8-10 octobre 1984.
43. Mattison HR, Reichman RC, Benedetti J, Bolgiano D, Davis LG, Bailey-Farchione A et al. Essai en double aveugle contrôlé par placebo comparant un traitement suppressif à long terme à un traitement à court terme par acyclovir oral pour la prise en charge de l'herpès génital récurrent. Am J Med 1988; 85(2A):20-25.
44. McLaren C, Sibrack CD, Barry DW. Spectre de sensibilité de l'acyclovir des isolats cliniques du virus de l'herpès simplex. Am J Med 1982; 73(1A):376-379.
45. McLaren C, Ellis MN, Hunter GA. Un test colorimétrique pour la mesure de la sensibilité des virus de l'herpès simplex aux agents antiviraux. Antiviral Res 1983; 3(4):223-234.
46. McLaren C, Corey L, Dekket C, Barry DW. Sensibilité in vitro à l'acyclovir dans les virus de l'herpès génital chez des patients traités à l'acyclovir. J Infect Dis 1983; 148(5):868-875.
47. Mertz GJ, Critchlow CW, Benedetti J, Reichman RC, Dolin R, Connor J et al. Essai contrôlé par placebo en double aveugle sur l'acyclovir oral dans le premier épisode d'infection par le virus de l'herpès simplex génital. JAMA 1984; 252(9):1147-1151.
48. Mertz GJ, Jones CC, Mills J, Fife KH, Lemon SM, Stapleton JT et al. Suppression à long terme de l'acyclovir de l'infection par le virus de l'herpès simplex génital fréquemment récurrente. Un essai multicentrique en double aveugle. JAMA 1988; 260(2):201-206.
49. Mertz GJ, Eron L, Kaufman R, Goldberg L, Raab B, Conant M et al. Traitement continu prolongé par rapport à l'acyclovir oral intermittent chez des adultes normaux atteints d'une infection par le virus de l'herpès simplex génital fréquemment récurrente. Am J Med 1988; 85(2A):14-19.
50. Meyer LJ, de Miranda P, Sheth N, Spruance S. Acyclovir dans le lait maternel humain. Am J Obstet Gynecol 1988; 158(3 Pt 1):586-588.
51. Mindel A, Weller IV, Faherty A, Sutherland S, Hindley D, Fiddian AP et al. Acyclovir oral prophylactique dans l'herpès génital récurrent. Lancette 1984 ; 2(8394):57-59.
52. Morton P, Thomson AN. Acyclovir oral dans le traitement du zona en médecine générale. NZ Med J 1989; 102(863):93-95.
53. Naib ZM, Nahmias AJ, Josey WE, Zaki SA. Relation de la cytohistopathologie de l'infection par le virus de l'herpès génital à l'anaplasie cervicale. Cancer Res 1973; 33(6):1452-1463.
54. Nilsen AE, Aasen T, Halsos AM, Kinge BR, Tjotta EA, Wikstrom K et al. Efficacité de l'acyclovir oral dans le traitement de l'herpès génital initial et récurrent. Lancette 1982 ; 2(8298):571-573.
55. Nusinoff-Lehrman S, Hunter G, Rogers J, Corey L, Davis G. La sensibilité in vitro à l'acyclovir de l'herpèsvirus excrété par les patients recevant une thérapie orale suppressive. 24th Interscience Conf Antimicrob Ag Chemother, Washington, DC, 8-10 octobre 1984 ; (Résumé #1015).
56. O'Brien JJ, Campoli-Richards DM. Acyclovir. Une revue mise à jour de son activité antivirale, de ses propriétés pharmacocinétiques et de son efficacité thérapeutique. Drogues 1989 ; 37(3):233-309.
57. Pahwa S, Biron K, Lim W, Swenson P, Kaplan MH, Sadick N et al. Infection varicelle-zona continue associée à une résistance à l'acyclovir chez un enfant atteint du SIDA. JAMA 1988; 260(19):2879-2882.
58. Parker AC, Craig JI, Collins P, Oliver N, Smith I. Infection par le virus de l'herpès simplex résistant à l'acyclovir due à une ADN polymérase altérée. Lancette 1987 ; 2(8573):1461.
59. Parris DS, Harrington JE. Des variantes du virus de l'herpès simplex limitées à des concentrations élevées d'acyclovir existent dans des isolats cliniques. Agents antimicrobiens Chemother 1982 ; 22(1):71-77.
60. Preblud SR, Arbeter AM, Proctor EA, Starr SE, Plotkin SA. Sensibilité des souches vaccinales du virus varicelle-zona aux composés antiviraux. Agents antimicrobiens Chemother 1984 ; 25(4):417-421.
61. Reichman RC, Badger GJ, Mertz GJ, Corey L, Richman DD, Connor JD et al. Traitement des infections génitales récurrentes à herpès simplex par l'acyclovir par voie orale. Un procès contrôlé. JAMA 1984; 251(16):2103-2107.
62. Shah GM, Winer RL, Krasny HC. Pharmacocinétique de l'acyclovir chez un patient sous dialyse péritonéale continue ambulatoire. Suis J Kidney Dis 1986; 7(6):507-510.
63. Sibrack CD, Gutman LT, Wilfert CM, McLaren C, St Clair MH, Keller PM et al. Pathogénicité du virus de l'herpès simplex de type 1 résistant à l'acyclovir chez un enfant immunodéprimé. J Infect Dis 1982; 146(5):673-682.
64. Straus SE, Seidlin M, Takiff H, Jacobs D, Bowen D, Smith HA. Acyclovir oral pour supprimer les infections récurrentes par le virus de l'herpès simplex chez les patients immunodéprimés. Ann Intern Med 1984 ; 100(4):522-524.
65. Straus SE, Takiff HE, Seidlin M, Bachrach S, Lininger L, DiGiovanna JJ et al. Suppression de l'herpès génital fréquemment récurrent. Un essai en double aveugle contrôlé par placebo sur l'acyclovir oral. N Engl J Med 1984; 310(24):1545-1550.
66. Straus SE, Croen KD, Sawyer MH, Freifeld AG, Felser JM, Dale JK et al. Suppression par l'acyclovir de l'herpès génital fréquemment récurrent. Efficacité et diminution des besoins au cours des années successives de traitement. JAMA 1988; 260(15):2227-2230.
67. Vinckier F, Boogaerts M, De Clerck D, De Clercq E. Infection herpétique chronique chez un patient immunodéprimé : rapport d'un cas. J Oral Maxillofac Surg 1987 ; 45(8):723-728.
68. Wade JC, Newton B, McLaren C, Flournoy N, Keeney RE, Meyers JD. Acyclovir intraveineux pour traiter l'infection mucocutanée par le virus de l'herpès simplex après une greffe de moelle: un essai en double aveugle. Ann Intern Med 1982 ; 96(3):265-269.
69. Wade JC, McLaren C, Meyers JD. Fréquence et importance du virus de l'herpès simplex résistant à l'acyclovir isolé chez des patients transplantés de moelle osseuse recevant plusieurs cycles de traitement à l'acyclovir. J Infect Dis 1983; 148(6):1077-1082.
INFORMATIONS PATIENTS
Les patientes sont invitées à consulter leur médecin si elles présentent des effets indésirables graves ou gênants, si elles tombent enceintes ou ont l'intention de le devenir, si elles ont l'intention d'allaiter tout en prenant ZOVIRAX (acyclovir) administré par voie orale ou si elles ont d'autres questions.
Il faut conseiller aux patients de maintenir une hydratation adéquate.