Epivir 150mg Lamivudine Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce qu'Epivir et comment est-il utilisé ?
Epivir 150 mg est un médicament sur ordonnance utilisé pour traiter les symptômes de l'infection par le VIH et de l'hépatite B chronique. Epivir 150 mg peut être utilisé seul ou avec d'autres médicaments.
Epivir appartient à une classe de médicaments appelés agents de l'hépatite B/hépatite C ; VIH, INTI.
On ne sait pas si Epivir est sûr et efficace chez les enfants de moins de 3 mois.
Quels sont les effets secondaires possibles d'Epivir 150 mg ?
Epivir peut provoquer des effets secondaires graves, notamment :
- urticaire,
- difficulté à respirer,
- gonflement du visage, des lèvres, de la langue ou de la gorge,
- douleurs musculaires inhabituelles,
- Douleur d'estomac,
- vomissement,
- rythme cardiaque irrégulier,
- vertiges,
- avoir froid,
- la faiblesse,
- fatigue,
- douleur intense dans le haut de l'estomac se propageant à votre dos,
- nausée,
- rythme cardiaque rapide,
- gonflement autour de votre abdomen,
- douleur dans le haut du ventre droit,
- perte d'appétit,
- urine foncée,
- selles de couleur argile,
- jaunissement de la peau ou des yeux (jaunisse),
- fièvre,
- sueurs nocturnes,
- glandes enflées,
- boutons de fièvre,
- toux,
- respiration sifflante,
- diarrhée,
- perte de poids,
- difficulté à parler ou à avaler,
- problèmes d'équilibre ou de mouvement des yeux,
- sensation de picotement,
- gonflement du cou ou de la gorge (hypertrophie de la thyroïde),
- changements menstruels, et
- impuissance
Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires les plus courants d'Epivir comprennent :
- nausée,
- diarrhée,
- mal de tête,
- fièvre,
- fatigue,
- malaise général,
- douleur à l'oreille ou sensation de satiété,
- difficulté à entendre,
- drainage de l'oreille,
- agitation chez un enfant,
- nez encombré,
- éternuements,
- mal de gorge, et
- toux
Dites au médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles d'Epivir. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
ATTENTION
EXACERBATIONS DE L'HÉPATITE B, ET DIFFÉRENTES FORMULATIONS D'EPIVIR.
Exacerbations de l'hépatite B
Des exacerbations aiguës sévères de l'hépatite B ont été rapportées chez des patients co-infectés par le virus de l'hépatite B (VHB) et le virus de l'immunodéficience humaine (VIH-1) et qui ont arrêté EPIVIR. La fonction hépatique doit être surveillée étroitement avec un suivi clinique et biologique pendant au moins plusieurs mois chez les patients qui arrêtent EPIVIR et qui sont co-infectés par le VIH-1 et le VHB. Le cas échéant, l'instauration d'un traitement anti-hépatite B peut être justifiée [voir MISES EN GARDE ET PRÉCAUTIONS].
Différences importantes entre les produits contenant de la lamivudine
Les comprimés et la solution buvable EPIVIR 150 mg (utilisés pour traiter l'infection par le VIH-1) contiennent une dose plus élevée de principe actif (lamivudine) que les comprimés et la solution buvable EPIVIR-HBV (utilisés pour traiter l'infection chronique par le VHB). Les patients infectés par le VIH-1 ne doivent recevoir que des formes posologiques appropriées pour le traitement du VIH-1 [voir MISES EN GARDE ET PRÉCAUTIONS].
LA DESCRIPTION
EPIVIR (également connu sous le nom de 3TC) est un nom de marque pour la lamivudine, un analogue nucléosidique synthétique ayant une activité contre le VIH-1 et le VHB. Le nom chimique de la lamivudine est (2R,cis)-4-amino-1(2-hydroxyméthyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidine-2-one. La lamivudine est l'énantiomère (-) d'un analogue didésoxy de la cytidine. La lamivudine a également été appelée (-)2',3'-didésoxy, 3'thiacytidine. Il a une formule moléculaire de C8H11N3O3S et un poids moléculaire de 229,3 g par mol. Il a la formule structurale suivante :
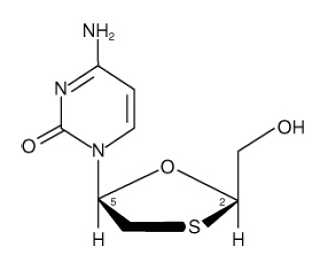
La lamivudine est un solide cristallin blanc à blanc cassé avec une solubilité d'environ 70 mg par mL dans l'eau à 20 °C.
Les comprimés d'EPIVIR 150 mg sont destinés à une administration orale. Chaque comprimé pelliculé sécable de 150 mg contient 150 mg de lamivudine et les ingrédients inactifs hypromellose, stéarate de magnésium, cellulose microcristalline, polyéthylèneglycol, polysorbate 80, glycolate d'amidon sodique et dioxyde de titane.
Chaque comprimé pelliculé de 300 mg contient 300 mg de lamivudine et les ingrédients inactifs oxyde de fer noir, hypromellose, stéarate de magnésium, cellulose microcristalline, polyéthylèneglycol, polysorbate 80, glycolate d'amidon sodique et dioxyde de titane.
EPIVIR solution buvable est destiné à une administration orale. Un millilitre (1 ml) de solution buvable EPIVIR contient 10 mg de lamivudine (10 mg par ml) dans une solution aqueuse et les ingrédients inactifs arômes artificiels de fraise et de banane, acide citrique (anhydre), méthylparabène, propylène glycol, propylparabène, citrate de sodium (dihydraté) et saccharose (200 mg).
LES INDICATIONS
EPIVIR 150 mg est un analogue nucléosidique indiqué en association avec d'autres agents antirétroviraux pour le traitement de l'infection par le virus de l'immunodéficience humaine de type 1 (VIH-1).
Limites d'utilisation
- Le dosage de ce produit est pour le VIH-1 et non pour le VHB.
DOSAGE ET ADMINISTRATION
Posologie recommandée pour les patients adultes
La posologie recommandée d'EPIVIR 150 mg chez les adultes infectés par le VIH-1 est de 300 mg par jour, administrée sous forme de 150 mg pris par voie orale deux fois par jour ou de 300 mg pris par voie orale une fois par jour avec ou sans nourriture. Si la lamivudine est administrée à un patient infecté par le VIH-1 et le VHB, la posologie indiquée pour le traitement du VIH-1 doit être utilisée dans le cadre d'un schéma thérapeutique combiné approprié [voir AVERTISSEMENTS ET PRECAUTIONS ].
Posologie recommandée pour les patients pédiatriques
EPIVIR 150 mg, comprimé sécable est la formulation préférée des patients pédiatriques infectés par le VIH-1 pesant au moins 14 kg et pour lesquels une forme galénique solide est appropriée. Avant de prescrire des comprimés sécables EPIVIR, les patients pédiatriques doivent être évalués pour leur capacité à avaler des comprimés. Pour les patients incapables d'avaler les comprimés d'EPIVIR 150 mg de manière sûre et fiable, la formulation en solution buvable peut être prescrite [voir AVERTISSEMENTS ET PRECAUTIONS ]. La posologie orale recommandée des comprimés EPIVIR 150 mg pour les patients pédiatriques infectés par le VIH-1 est présentée dans le tableau 1.
Solution orale
La posologie recommandée d'EPIVIR 150 mg solution buvable chez les patients pédiatriques infectés par le VIH-1 âgés de 3 mois et plus est de 5 mg par kg pris par voie orale deux fois par jour ou de 10 mg par kg pris par voie orale une fois par jour (jusqu'à un maximum de 300 mg par jour), administré en association avec d'autres agents antirétroviraux [voir PHARMACOLOGIE CLINIQUE ]. Tenez compte de la charge virale du VIH-1 et de la numération/du pourcentage de cellules CD4+ lors de la sélection de l'intervalle de dosage pour les patients débutant un traitement avec une solution buvable [voir AVERTISSEMENTS ET PRECAUTIONS , PHARMACOLOGIE CLINIQUE ].
Patients atteints d'insuffisance rénale
La posologie d'EPIVIR 150 mg est adaptée en fonction de la fonction rénale. Les ajustements posologiques sont répertoriés dans le tableau 2 [voir PHARMACOLOGIE CLINIQUE ].
Aucune dose supplémentaire d'EPIVIR 150 mg n'est nécessaire après une hémodialyse de routine (4 heures) ou une dialyse péritonéale.
Bien qu'il n'y ait pas suffisamment de données pour recommander un ajustement de dose spécifique d'EPIVIR chez les patients pédiatriques atteints d'insuffisance rénale, une réduction de la dose et/ou une augmentation de l'intervalle de dosage doit être envisagée.
COMMENT FOURNIE
Formes posologiques et points forts
EPIVIR Comprimés Sécables
Les comprimés sécables EPIVIR contiennent 150 mg de lamivudine. Les comprimés sont blancs, en forme de losange, sécables, pelliculés et portant l'inscription « GX CJ7 » gravée sur les deux faces.
Comprimés EPIVIR
Les comprimés EPIVIR contiennent 300 mg de lamivudine. Les comprimés sont gris, en forme de losange modifié, pelliculés et gravés « GX EJ7 » sur une face et lisses sur l'autre face.
EPIVIR Solution buvable
La solution buvable EPIVIR contient 10 mg de lamivudine pour 1 mL. La solution est un liquide clair, incolore à jaune pâle, à saveur de fraise-banane.
Stockage et manutention
ÉPIVIR Les comprimés sécables contiennent 150 mg de lamivudine, sont blancs, en forme de losange, pelliculés et gravés « GX CJ7 » des deux côtés. Emballé comme suit :
Flacon de 60 comprimés ( CDN 49702-203-18) avec fermeture à l'épreuve des enfants.
ÉPIVIR Les comprimés contiennent 300 mg de lamivudine, sont gris, en forme de losange modifié, pelliculés et gravés « GX EJ7 » sur une face et lisses sur l'autre face. Emballé comme suit :
Flacon de 30 comprimés ( CDN 49702-204-13) avec fermeture à l'épreuve des enfants.
Stockage recommandé
Conservez les comprimés EPIVIR à 25°C (77°F); excursions permises jusqu'à 15° à 30°C (59° à 86°F) [voir Température ambiante contrôlée USP ].
EPIVIR solution buvable est un liquide limpide, incolore à jaune pâle, au goût fraise-banane. Chaque mL de solution contient 10 mg de lamivudine. Emballé comme suit :
Bouteille en plastique de 240 ml (NDC 49702-205-48) avec fermeture à l'épreuve des enfants. Ce produit ne nécessite pas de reconstitution.
Conserver dans des flacons hermétiquement fermés à 25 °C (77 °F) [voir Température ambiante contrôlée USP ].
Fabriqué pour : ViiV Healthcare Research Triangle Park, NC 27709. par : GlaxoSmithKline Research Triangle Park, NC 27709, Fabriqué sous accord de Shire Pharmaceuticals Group plc. Révisé : avril 2018
EFFETS SECONDAIRES
Les effets indésirables suivants sont discutés dans d'autres sections de l'étiquetage :
- Exacerbations de l'hépatite B [voir AVERTISSEMENT ENCADRÉ , AVERTISSEMENTS ET PRECAUTIONS ].
- Acidose lactique et hépatomégalie sévère avec stéatose [voir AVERTISSEMENTS ET PRECAUTIONS ].
- Décompensation hépatique chez les patients co-infectés par le VIH-1 et l'hépatite C [voir AVERTISSEMENTS ET PRECAUTIONS ].
- Pancréatite [voir AVERTISSEMENTS ET PRECAUTIONS ].
- Syndrome de reconstitution immunitaire [voir AVERTISSEMENTS ET PRECAUTIONS ].
Expérience des essais cliniques
Expérience d'essais cliniques chez des sujets adultes
Étant donné que les essais cliniques sont menés dans des conditions très variables, les taux d'effets indésirables observés dans les essais cliniques d'un médicament ne peuvent pas être directement comparés aux taux des essais cliniques d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique clinique.
Le profil de sécurité d'EPIVIR 150 mg chez l'adulte est principalement basé sur 3 568 sujets infectés par le VIH-1 dans 7 essais cliniques.
Les effets indésirables les plus courants sont les maux de tête, les nausées, les malaises, la fatigue, les signes et symptômes nasaux, la diarrhée et la toux.
Les réactions défavorables cliniques choisies dans plus grand qu'ou égal à 5 % de sujets pendant la thérapie avec EPIVIR 150 mgs deux fois par jour plus RETROVIR 200 mgs 3 fois tous les jours depuis jusqu'à 24 semaines sont énumérées dans la Table 3.
Pancréatite
Une pancréatite a été observée chez 9 des 2 613 sujets adultes (0,3 %) ayant reçu EPIVIR 150 mg dans les essais cliniques contrôlés EPV20001, NUCA3001, NUCB3001, NUCA3002, NUCB3002 et NUCB3007 [voir AVERTISSEMENTS ET PRECAUTIONS ].
EPIVIR 300 mg une fois par jour
Les types et les fréquences de réactions défavorables cliniques ont annoncé dans les sujets recevant EPIVIR 300 mgs une fois tous les jours ou EPIVIR 150 mgs deux fois par jour (dans les régimes de combinaison de 3 médicaments dans EPV20001 et EPV40001) depuis 48 semaines étaient semblables.
Les anomalies de laboratoire sélectionnées observées pendant le traitement sont résumées dans le tableau 4.
Les fréquences d'anomalies de laboratoire choisies annoncées dans les sujets recevant EPIVIR 300 mgs une fois tous les jours ou EPIVIR 150 mgs deux fois par jour (dans les régimes de combinaison de 3 médicaments dans EPV20001 et EPV40001) étaient semblables.
Expérience d'essais cliniques chez des sujets pédiatriques
EPIVIR solution buvable a été étudié chez 638 sujets pédiatriques âgés de 3 mois à 18 ans dans 3 essais cliniques.
Effets indésirables cliniques sélectionnés et signes physiques avec une fréquence supérieure ou égale à 5 % pendant le traitement par EPIVIR 4 mg par kg deux fois par jour plus RETROVIR 160 mg par m² 3 fois par jour chez les patients naïfs de traitement (moins de ou égal à 56 jours de traitement antirétroviral). thérapie) les sujets pédiatriques sont répertoriés dans le tableau 5.
Pancréatite
La pancréatite, qui a été fatale dans certains cas, a été observée chez des sujets pédiatriques ayant déjà été traités par des nucléosides antirétroviraux recevant EPIVIR seul ou en association avec d'autres agents antirétroviraux. Dans un procès d'escalade de dose d'étiquette ouverte (NUCA2002), 14 sujets (14 %) ont développé la pancréatite en recevant la monothérapie avec EPIVIR. Trois de ces sujets sont décédés de complications de pancréatite. Dans un deuxième essai ouvert (NUCA2005), 12 sujets (18 %) ont développé une pancréatite. Dans le Procès ACTG300, la pancréatite n'a pas été observée dans 236 sujets randomisés à EPIVIR 150 mgs plus RETROVIR. Une pancréatite a été observée chez 1 sujet de cet essai qui a reçu EPIVIR en ouvert en association avec RETROVIR et ritonavir après l'arrêt de la didanosine en monothérapie [voir AVERTISSEMENTS ET PRECAUTIONS ].
Paresthésies et neuropathies périphériques
Des paresthésies et des neuropathies périphériques ont été signalées chez 15 sujets (15 %) dans l'essai NUCA2002, 6 sujets (9 %) dans l'essai NUCA2005 et 2 sujets (moins de 1 %) dans l'essai ACTG300.
Les anomalies de laboratoire sélectionnées rencontrées par les sujets pédiatriques naïfs de traitement (moins de ou égal à 56 jours de traitement antirétroviral) sont répertoriées dans le tableau 6.
Sujets pédiatriques Dosage une fois par jour ou deux fois par jour (COL105677)
L'innocuité de l'administration uniquotidienne par rapport à l'administration biquotidienne d'EPIVIR 150 mg a été évaluée dans l'essai ARROW. L'évaluation primaire de l'innocuité dans l'essai ARROW était basée sur les événements indésirables de grade 3 et de grade 4. La fréquence des événements indésirables de grade 3 et 4 était similaire chez les sujets randomisés pour une dose uniquotidienne par rapport aux sujets randomisés pour une dose biquotidienne. Un événement d'hépatite de grade 4 dans la cohorte à prise unique quotidienne a été considéré comme étant de causalité incertaine par l'investigateur et tous les autres événements indésirables de grade 3 ou 4 ont été considérés comme non liés par l'investigateur.
Nouveau-nés
Des informations limitées sur l'innocuité à court terme sont disponibles à partir de 2 petits essais non contrôlés en Afrique du Sud chez des nouveau-nés recevant de la lamivudine avec ou sans zidovudine pendant la première semaine de vie après un traitement maternel commençant à la semaine 38 ou 36 de la gestation [voir PHARMACOLOGIE CLINIQUE ]. Les effets indésirables sélectionnés signalés chez ces nouveau-nés comprenaient une augmentation des tests de la fonction hépatique, l'anémie, la diarrhée, les troubles électrolytiques, l'hypoglycémie, la jaunisse et l'hépatomégalie, les éruptions cutanées, les infections respiratoires et la septicémie ; 3 nouveau-nés sont décédés (1 de gastro-entérite avec acidose et convulsions, 1 de blessure traumatique et 1 de causes inconnues). Deux autres cas de gastro-entérite ou de diarrhée non fatals ont été signalés, dont 1 avec convulsions ; 1 enfant avait une insuffisance rénale transitoire associée à une déshydratation. L'absence de groupes témoins limite les évaluations de la causalité, mais il faut supposer que les nourrissons exposés pendant la période périnatale peuvent présenter un risque d'effets indésirables comparables à ceux rapportés chez les patients pédiatriques et adultes infectés par le VIH-1 traités par des combinaisons thérapeutiques contenant de la lamivudine. Les effets à long terme de l'exposition in utero et infantile à la lamivudine ne sont pas connus.
Expérience post-commercialisation
Les réactions défavorables suivantes ont été identifiées pendant l'utilisation de post-approbation d'EPIVIR. Étant donné que ces réactions sont signalées volontairement par une population de taille inconnue, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale avec l'exposition au médicament. Ces réactions ont été choisies pour inclusion en raison d'une combinaison de leur gravité, de leur fréquence de notification ou de leur lien de causalité potentiel avec la lamivudine.
Corps dans son ensemble
Redistribution/accumulation de graisse corporelle.
Endocrinien et Métabolique
Hyperglycémie.
Général
La faiblesse. Anémie hémique et lymphatique (y compris l'aplasie pure des globules rouges et les anémies sévères progressant sous traitement).
Hépatique et pancréatique
Acidose lactique et stéatose hépatique [voir AVERTISSEMENTS ET PRECAUTIONS ], exacerbations post-traitement de l'hépatite B [voir AVERTISSEMENTS ET PRECAUTIONS ].
Hypersensibilité
Anaphylaxie, urticaire.
Musculo-squelettique
Faiblesse musculaire, élévation des CPK, rhabdomyolyse. Peau Alopécie, prurit.
INTERACTIONS MÉDICAMENTEUSES
Médicaments inhibant les transporteurs de cations organiques
La lamivudine est principalement éliminée dans les urines par sécrétion cationique organique active. La possibilité d'interactions avec d'autres médicaments administrés simultanément doit être envisagée, en particulier lorsque leur principale voie d'élimination est la sécrétion rénale active via le système de transport cationique organique (par exemple, le triméthoprime) [voir PHARMACOLOGIE CLINIQUE ]. Aucune donnée n'est disponible concernant les interactions avec d'autres médicaments ayant des mécanismes de clairance rénale similaires à ceux de la lamivudine.
Sorbitol
L'administration concomitante de doses uniques de lamivudine et de sorbitol a entraîné une réduction dose-dépendante du sorbitol des expositions à la lamivudine. Dans la mesure du possible, évitez d'utiliser des médicaments contenant du sorbitol avec la lamivudine [voir AVERTISSEMENTS ET PRECAUTIONS , PHARMACOLOGIE CLINIQUE ].
AVERTISSEMENTS
Inclus dans le cadre du PRÉCAUTIONS section.
PRÉCAUTIONS
Patients co-infectés par le virus de l'hépatite B
Exacerbations post-traitement de l'hépatite
Des preuves cliniques et de laboratoire d'exacerbations d'hépatite sont survenues après l'arrêt de la lamivudine. Ces exacerbations ont été détectées principalement par des élévations d'ALT sérique en plus de la réémergence de l'ADN du VHB. Bien que la plupart des événements semblent avoir été spontanément résolutifs, des décès ont été signalés dans certains cas. Des événements similaires ont été rapportés depuis la commercialisation après le passage de schémas thérapeutiques anti-VIH-1 contenant de la lamivudine à des schémas thérapeutiques ne contenant pas de lamivudine chez des patients infectés à la fois par le VIH-1 et le VHB. La relation causale avec l'arrêt du traitement par la lamivudine n'est pas connue. Les patients doivent être étroitement surveillés avec un suivi clinique et biologique pendant au moins plusieurs mois après l'arrêt du traitement.
Différences importantes entre les produits contenant de la lamivudine
Les comprimés et la solution buvable EPIVIR 150 mg contiennent une dose plus élevée du même principe actif (lamivudine) que les comprimés EPIVIR-HBV et la solution buvable EPIVIR-HBV. EPIVIR-HBV a été développé pour les patients atteints d'hépatite B chronique. La formulation et la posologie de la lamivudine dans EPIVIR-HBV ne sont pas appropriées pour les patients co-infectés par le VIH-1 et le VHB. L'innocuité et l'efficacité de la lamivudine n'ont pas été établies pour le traitement de l'hépatite B chronique chez les patients co-infectés par le VIH-1 et le VHB. Si un traitement par EPIVIR-HBV est prescrit pour l'hépatite B chronique chez un patient atteint d'une infection par le VIH-1 non reconnue ou non traitée, l'émergence rapide d'une résistance au VIH-1 est susceptible de se produire en raison de la dose sous-thérapeutique et de l'inadéquation du traitement du VIH-1 en monothérapie. S'il est décidé d'administrer la lamivudine à des patients co-infectés par le VIH-1 et le VHB, les comprimés EPIVIR 150 mg, EPIVIR 150 mg solution buvable ou un autre produit contenant la dose la plus élevée de lamivudine doivent être utilisés dans le cadre d'un schéma thérapeutique combiné approprié.
Émergence du VHB résistant à la lamivudine
La sécurité et l'efficacité de la lamivudine n'ont pas été établies pour le traitement de l'hépatite B chronique chez les sujets doublement infectés par le VIH-1 et le VHB (voir les informations de prescription complètes pour EPIVIR-HBV). L'émergence de variants du virus de l'hépatite B associée à une résistance à la lamivudine a également été rapportée chez des sujets infectés par le VIH-1 ayant reçu des traitements antirétroviraux contenant de la lamivudine en présence d'une infection concomitante par le virus de l'hépatite B.
Acidose lactique et hépatomégalie sévère avec stéatose
Une acidose lactique et une hépatomégalie sévère avec stéatose, y compris des cas mortels, ont été rapportées avec l'utilisation d'analogues nucléosidiques, dont EPIVIR. La majorité de ces cas concernaient des femmes. Le sexe féminin et l'obésité peuvent être des facteurs de risque de développement d'acidose lactique et d'hépatomégalie sévère avec stéatose chez les patients traités par analogues nucléosidiques antirétroviraux. Le traitement par EPIVIR doit être suspendu chez tout patient qui développe des signes cliniques ou de laboratoire suggérant une acidose lactique ou une hépatotoxicité prononcée, pouvant inclure une hépatomégalie et une stéatose même en l'absence d'élévations marquées des transaminases.
Utiliser avec les régimes à base d'interféron et de ribavirine
Des études in vitro ont montré que la ribavirine peut réduire la phosphorylation des analogues nucléosidiques de la pyrimidine tels que la lamivudine. Bien qu'aucune preuve d'interaction pharmacocinétique ou pharmacodynamique (p. ex., perte de la suppression virologique du VIH-1/VHC) n'ait été observée lorsque la ribavirine était co-administrée avec la lamivudine chez des patients co-infectés par le VIH-1/VHC [voir PHARMACOLOGIE CLINIQUE ], une décompensation hépatique (certaines mortelles) est survenue chez des patients co-infectés par le VIH-1/VHC recevant une thérapie antirétrovirale combinée pour le VIH-1 et l'interféron alfa avec ou sans ribavirine. Les patients recevant de l'interféron alfa avec ou sans ribavirine et EPIVIR doivent être étroitement surveillés pour les toxicités associées au traitement, en particulier la décompensation hépatique. L'arrêt d'EPIVIR 150 mg doit être considéré comme médicalement approprié. Une réduction de la dose ou l'arrêt de l'interféron alfa, de la ribavirine ou des deux doivent également être envisagés si une aggravation des toxicités cliniques est observée, y compris une décompensation hépatique (p. ex., Child-Pugh supérieur à 6). Voir les informations de prescription complètes pour l'interféron et la ribavirine.
Pancréatite
Chez les patients pédiatriques ayant des antécédents d'exposition antérieure aux nucléosides antirétroviraux, des antécédents de pancréatite ou d'autres facteurs de risque significatifs de développement de pancréatite, EPIVIR 150 mg doit être utilisé avec prudence. Le traitement par EPIVIR 150 mg doit être arrêté immédiatement si des signes cliniques, des symptômes ou des anomalies de laboratoire évoquant une pancréatite surviennent [voir EFFETS INDÉSIRABLES ].
Syndrome de reconstitution immunitaire
Un syndrome de reconstitution immunitaire a été rapporté chez des patients traités par une thérapie antirétrovirale combinée, y compris EPIVIR. Au cours de la phase initiale du traitement antirétroviral combiné, les patients dont le système immunitaire répond peuvent développer une réponse inflammatoire aux infections opportunistes indolentes ou résiduelles (telles que l'infection à Mycobacterium avium, le cytomégalovirus, la pneumonie à Pneumocystis jirovecii [PCP] ou la tuberculose), ce qui peut nécessiter une évaluation plus approfondie et traitement.
Des troubles auto-immuns (tels que la maladie de Graves, la polymyosite et le syndrome de Guillain-Barré) ont également été signalés dans le cadre de la reconstitution immunitaire, cependant, le délai d'apparition est plus variable et peut survenir plusieurs mois après le début du traitement.
Taux de suppression virologique inférieurs et risque accru de résistance virale avec la solution orale
Les sujets pédiatriques qui ont reçu EPIVIR 150 mg solution buvable (à des doses basées sur la tranche de poids d'environ 8 mg par kg par jour) en concomitance avec d'autres solutions buvables antirétrovirales à tout moment de l'essai ARROW ont présenté des taux plus faibles de suppression virologique, une exposition plasmatique à la lamivudine plus faible et ont développé résistance virale plus fréquemment que ceux recevant des comprimés EPIVIR [voir PHARMACOLOGIE CLINIQUE , Microbiologie , Etudes cliniques ].
EPIVIR comprimé sécable est la formulation préférée des patients pédiatriques infectés par le VIH-1 qui pèsent au moins 14 kg et pour lesquels une forme posologique solide est appropriée. Dans la mesure du possible, un régime à base de comprimés doit être utilisé pour éviter une interaction potentielle avec le sorbitol [voir PHARMACOLOGIE CLINIQUE ]. Envisager une surveillance plus fréquente de la charge virale VIH-1 lors du traitement par EPIVIR 150 mg solution buvable.
Informations sur les conseils aux patients
Conseillez au patient de lire l'étiquetage patient approuvé par la FDA (Informations patient).
Patients co-infectés par l'hépatite B ou C
Informez les patients co-infectés par le VIH-1 et le VHB qu'une détérioration de la maladie hépatique s'est produite dans certains cas lorsque le traitement par la lamivudine a été interrompu. Conseillez aux patients de discuter de tout changement de régime avec leur fournisseur de soins de santé [voir AVERTISSEMENTS ET PRECAUTIONS ].
Informer les patients co-infectés par le VIH-1/VHC qu'une décompensation hépatique (certaine fatale) s'est produite chez les patients co-infectés par le VIH-1/VHC recevant une thérapie antirétrovirale combinée pour le VIH-1 et l'interféron alfa avec ou sans ribavirine [voir AVERTISSEMENTS ET PRECAUTIONS ].
Différences dans les formulations d'EPIVIR
Aviser les patients qu'EPIVIR 150 mg comprimés et solution buvable contiennent une dose plus élevée du même ingrédient actif (lamivudine) que les comprimés EPIVIR-HBV et la solution buvable. Si une décision est prise d'inclure la lamivudine dans le schéma thérapeutique du VIH-1 d'un patient co-infecté par le VIH-1 et le VHB, la formulation et la posologie de la lamivudine dans EPIVIR (et non EPIVIR-VHB) doivent être utilisées [voir AVERTISSEMENTS ET PRECAUTIONS ].
Acidose lactique/hépatomégalie avec stéatose
Informer les patients qu'une acidose lactique et une hépatomégalie sévère avec stéatose ont été rapportées avec l'utilisation d'analogues nucléosidiques et d'autres antirétroviraux. Conseillez aux patients d'arrêter de prendre EPIVIR s'ils développent des symptômes cliniques évoquant une acidose lactique ou une hépatotoxicité prononcée [voir AVERTISSEMENTS ET PRECAUTIONS ].
Risque de pancréatite
Conseillez aux parents ou aux tuteurs de surveiller les patients pédiatriques pour détecter tout signe et symptôme de pancréatite [voir AVERTISSEMENTS ET PRECAUTIONS ].
Syndrome de reconstitution immunitaire
Conseillez aux patients d'informer immédiatement leur fournisseur de soins de santé de tout signe et symptôme d'infection car l'inflammation d'une infection antérieure peut survenir peu de temps après la thérapie antirétrovirale combinée, y compris lorsqu'EPIVIR est commencé [voir AVERTISSEMENTS ET PRECAUTIONS ].
Taux de suppression virologique inférieurs et risque accru de résistance virale avec la solution orale
Aviser les patients qu'un schéma posologique tout-comprimé doit être utilisé lorsque cela est possible en raison d'un taux accru d'échec du traitement chez les sujets pédiatriques qui ont reçu EPIVIR en solution orale en concomitance avec d'autres solutions orales antirétrovirales [voir AVERTISSEMENTS ET PRECAUTIONS ].
Teneur en saccharose de la solution buvable EPIVIR
Avisez les patients diabétiques que chaque dose de 15 ml de solution buvable EPIVIR contient 3 grammes de saccharose (1 ml = 200 mg de saccharose) [voir LA DESCRIPTION ].
Registre des grossesses
Avisez des patients qu'il y a un registre d'exposition de grossesse qui surveille des résultats de grossesse dans les femmes exposées à EPIVIR pendant la grossesse [voir Utilisation dans des populations spécifiques ].
Lactation
Demandez aux femmes infectées par le VIH-1 de ne pas allaiter car le VIH-1 peut être transmis au bébé par le lait maternel [voir Utilisation dans des populations spécifiques ].
Dosage oublié
Donnez l'ordre aux patients que s'ils manquent une dose d'EPIVIR 150 mgs, de le prendre dès qu'ils se rappellent. Conseillez aux patients de ne pas doubler leur dose suivante ou de prendre plus que la dose prescrite [voir DOSAGE ET ADMINISTRATION ].
EPIVIR et RETROVIR sont des marques déposées détenues ou utilisées sous licence par le groupe de sociétés ViiV Healthcare.
Toxicologie non clinique
Carcinogenèse, mutagenèse, altération de la fertilité
Carcinogenèse
Des études de cancérogénicité à long terme avec la lamivudine chez la souris et le rat n'ont montré aucun signe de potentiel carcinogène à des expositions jusqu'à 10 fois (souris) et 58 fois (rats) les expositions humaines à la dose recommandée de 300 mg.
Mutagenèse
La lamivudine s'est révélée mutagène dans un test de lymphome de souris L5178Y et clastogène dans un test cytogénétique utilisant des lymphocytes humains en culture. La lamivudine n'a pas été mutagène lors d'un test de mutagénicité microbienne, d'un test de transformation cellulaire in vitro, d'un test du micronoyau chez le rat, d'un test cytogénétique sur la moelle osseuse du rat et d'un test de synthèse d'ADN non programmée dans le foie du rat. La lamivudine n'a montré aucun signe d'activité génotoxique in vivo chez le rat à des doses orales allant jusqu'à 2 000 mg par kg, produisant des taux plasmatiques de 35 à 45 fois ceux chez l'homme à la dose recommandée pour l'infection par le VIH-1.
Altération de la fertilité
Dans une étude sur les performances de reproduction, la lamivudine administrée à des rats à des doses allant jusqu'à 4 000 mg par kg par jour, produisant des taux plasmatiques 47 à 70 fois supérieurs à ceux de l'homme, n'a révélé aucun signe d'altération de la fertilité et aucun effet sur la survie, la croissance et le développement. au sevrage de la progéniture.
Utilisation dans des populations spécifiques
Grossesse
Registre d'exposition pendant la grossesse
Il y a un enregistrement d'exposition de grossesse qui surveille des résultats de grossesse dans les femmes exposées à EPIVIR pendant la grossesse. Les prestataires de soins de santé sont encouragés à inscrire les patientes en appelant le registre des grossesses antirétrovirales (APR) au 1-800-258-4263.
Résumé des risques
Les données disponibles de l'APR ne montrent aucune différence dans le risque global de malformations congénitales pour la lamivudine par rapport au taux de fond de malformations congénitales de 2,7 % dans la population de référence du Metropolitan Atlanta Congenital Defects Program (MACDP) (voir Données ). L'APR utilise le MACDP comme population de référence américaine pour les malformations congénitales dans la population générale. Le MACDP évalue les femmes et les nourrissons d'une zone géographique limitée et n'inclut pas les résultats pour les naissances survenues à moins de 20 semaines de gestation. Le taux de fausses couches n'est pas rapporté dans l'APR. Le taux de fond estimé de fausse couche dans les grossesses cliniquement reconnues dans la population générale des États-Unis est de 15 % à 20 %. Le risque de fond de malformations congénitales majeures et de fausse couche pour la population indiquée est inconnu.
Dans les études de reproduction chez l'animal, l'administration orale de lamivudine à des lapines gestantes au cours de l'organogenèse a entraîné une embryolétalité à l'exposition systémique (ASC) similaire à la dose clinique recommandée ; cependant, aucun effet indésirable sur le développement n'a été observé avec l'administration orale de lamivudine à des rates gravides pendant l'organogenèse à des concentrations plasmatiques (Cmax) 35 fois la dose clinique recommandée (voir Données ).
Données
Données humaines
Sur la base des rapports prospectifs à l'APR de plus de 11 000 expositions à la lamivudine pendant la grossesse ayant entraîné des naissances vivantes (dont plus de 4 500 exposées au cours du premier trimestre), il n'y avait aucune différence entre le risque global d'anomalies congénitales pour la lamivudine et le taux d'anomalies congénitales de base de 2,7 % dans la population de référence américaine du MACDP. La prévalence des malformations chez les naissances vivantes était de 3,1 % (IC à 95 % : 2,6 % à 3,6 %) après l'exposition au premier trimestre à des schémas thérapeutiques contenant de la lamivudine et de 2,8 % (IC à 95 % : 2,5 % à 3,3 %) après l'exposition au deuxième/troisième trimestre. aux régimes contenant de la lamivudine.
La pharmacocinétique de la lamivudine a été étudiée chez des femmes enceintes au cours de 2 essais cliniques menés en Afrique du Sud. Les essais ont évalué la pharmacocinétique chez 16 femmes à 36 semaines de gestation utilisant 150 mg de lamivudine deux fois par jour avec de la zidovudine, 10 femmes à 38 semaines de gestation utilisant 150 mg de lamivudine deux fois par jour avec de la zidovudine et 10 femmes à 38 semaines de gestation utilisant la lamivudine 300 mg deux fois quotidiennement sans autres antirétroviraux. Ces essais n'ont pas été conçus ou alimentés pour fournir des informations sur l'efficacité. Les concentrations de lamivudine étaient généralement similaires dans les échantillons de sérum maternel, néonatal et de cordon ombilical. Dans un sous-ensemble de sujets, des échantillons de liquide amniotique ont été prélevés après une rupture naturelle des membranes et ont confirmé que la lamivudine traverse le placenta chez l'homme. Sur la base de données limitées à l'accouchement, les concentrations médianes (plage) de liquide amniotique de lamivudine étaient 3,9 (1,2 à 12,8) fois plus élevées que les concentrations sériques maternelles appariées (n = 8).
Données animales
La lamivudine a été administrée par voie orale à des rates gravides (à 90, 600 et 4 000 mg par kg par jour) et à des lapines (à 90, 300 et 1 000 mg par kg par jour et à 15, 40 et 90 mg par kg par jour) pendant l'organogenèse (les jours de gestation 7 à 16 [rat] et 8 à 20 [lapin]). Aucun signe de malformations fœtales dues à la lamivudine n'a été observé chez le rat et le lapin à des doses produisant des concentrations plasmatiques (Cmax) environ 35 fois supérieures à l'exposition humaine à la dose quotidienne recommandée. Des signes de létalité embryonnaire précoce ont été observés chez le lapin à des expositions systémiques (ASC) similaires à celles observées chez l'homme, mais il n'y avait aucune indication de cet effet chez le rat à des concentrations plasmatiques (Cmax) 35 fois supérieures à l'exposition humaine à la dose quotidienne recommandée. . Des études chez des rats gravides ont montré que la lamivudine est transférée au fœtus par le placenta. Dans l'étude de fertilité/développement prénatal et postnatal chez le rat, la lamivudine a été administrée par voie orale à des doses de 180, 900 et 4 000 mg par kg par jour (d'avant l'accouplement jusqu'au jour 20 postnatal). Dans l'étude, le développement de la progéniture, y compris la fertilité et la performance reproductive, n'a pas été affecté par l'administration maternelle de lamivudine.
Lactation
Résumé des risques
Les Centers for Disease Control and Prevention recommandent aux mères infectées par le VIH-1 aux États-Unis de ne pas allaiter leurs bébés pour éviter le risque de transmission postnatale de l'infection par le VIH-1. La lamivudine est présente dans le lait maternel. Il n'existe aucune information sur les effets de la lamivudine sur le nourrisson allaité ou sur les effets des médicaments sur la production de lait. En raison du risque de (1) transmission du VIH-1 (chez les nourrissons séronégatifs), (2) de développement d'une résistance virale (chez les nourrissons séropositifs) et (3) d'effets indésirables chez un nourrisson allaité, demandez aux mères de ne pas allaiter s'ils reçoivent EPIVIR.
Utilisation pédiatrique
La sécurité et l'efficacité d'EPIVIR 150 mg en association avec d'autres agents antirétroviraux ont été établies chez les patients pédiatriques âgés de 3 mois et plus. Le comprimé sécable EPIVIR est la formulation préférée pour les patients pédiatriques infectés par le VIH-1 qui pèsent au moins 14 kg et pour lesquels une forme posologique solide est appropriée parce que les sujets pédiatriques qui ont reçu EPIVIR 150 mg solution buvable présentaient des taux plus faibles de suppression virologique, une exposition plasmatique à la lamivudine plus faible , et ont développé une résistance virale plus fréquemment que ceux recevant des comprimés EPIVIR 150 mg dans l'essai ARROW [voir DOSAGE ET ADMINISTRATION , AVERTISSEMENTS ET PRECAUTIONS , EFFETS INDÉSIRABLES , PHARMACOLOGIE CLINIQUE , Etudes cliniques ].
Utilisation gériatrique
Les essais cliniques d'EPIVIR n'ont pas inclus un nombre suffisant de sujets âgés de 65 ans et plus pour déterminer s'ils répondent différemment des sujets plus jeunes. En général, la prudence devrait être exercée dans l'administration d'EPIVIR dans les patients assez âgés reflétant la plus grande fréquence de fonction hépatique, rénale, ou cardiaque diminuée et de maladie d'élément ou d'autre thérapie de médicament [voir DOSAGE ET ADMINISTRATION , PHARMACOLOGIE CLINIQUE ].
Patients atteints d'insuffisance rénale
Une réduction de la posologie d'EPIVIR 150 mg est recommandée chez les patients insuffisants rénaux [voir DOSAGE ET ADMINISTRATION , PHARMACOLOGIE CLINIQUE ].
SURDOSAGE
Il n'y a aucun traitement spécifique connu pour l'overdose avec EPIVIR. En cas de surdosage, le patient doit être surveillé et un traitement de soutien standard doit être appliqué si nécessaire. Étant donné qu'une quantité négligeable de lamivudine a été éliminée par hémodialyse (4 heures), dialyse péritonéale continue ambulatoire et dialyse péritonéale automatisée, on ne sait pas si l'hémodialyse continue apporterait un bénéfice clinique en cas de surdosage de lamivudine.
CONTRE-INDICATIONS
EPIVIR 150mg est contre-indiqué chez les patients ayant un antécédent de réaction d'hypersensibilité à la lamivudine.
PHARMACOLOGIE CLINIQUE
Mécanisme d'action
La lamivudine est un agent antirétroviral [voir Microbiologie ].
Pharmacocinétique
Pharmacocinétique chez l'adulte
Les propriétés pharmacocinétiques de la lamivudine ont été étudiées chez des sujets adultes asymptomatiques infectés par le VIH-1 après administration de doses intraveineuses (IV) uniques allant de 0,25 à 8 mg par kg, ainsi que de doses orales uniques et multiples (régime biquotidien). allant de 0,25 à 10 mg par kg.
Les propriétés pharmacocinétiques de la lamivudine ont également été étudiées sous forme de doses orales uniques et multiples allant de 5 mg à 600 mg par jour administrées à des sujets infectés par le VHB.
Les propriétés pharmacocinétiques à l'état d'équilibre du comprimé EPIVIR de 300 mg une fois par jour pendant 7 jours par rapport au comprimé EPIVIR de 150 mg deux fois par jour pendant 7 jours ont été évaluées dans un essai croisé chez 60 sujets sains. EPIVIR 300 mg une fois par jour a entraîné des expositions à la lamivudine qui étaient similaires à EPIVIR 150 mg deux fois par jour en ce qui concerne l'ASC24,ss plasmatique ; cependant, la Cmax,ss était supérieure de 66 % et la valeur minimale était inférieure de 53 % par rapport au régime de 150 mg deux fois par jour. Les expositions intracellulaires à la lamivudine triphosphate dans les cellules mononucléaires du sang périphérique étaient également similaires en ce qui concerne l'ASC24,ss et la Cmax24,ss ; cependant, les valeurs minimales étaient inférieures à celles du régime de 150 mg deux fois par jour. La variabilité interindividuelle était plus importante pour les concentrations intracellulaires de triphosphate de lamivudine que pour les concentrations plasmatiques minimales de lamivudine.
La pharmacocinétique de la lamivudine a été évaluée chez 12 sujets adultes infectés par le VIH-1 ayant reçu 150 mg de lamivudine deux fois par jour en association avec d'autres agents antirétroviraux. La moyenne géométrique (IC à 95 %) pour l'ASC(0-12) était de 5,53 (4,58, 6,67) mcg.h par mL et pour la Cmax était de 1,40 (1,17, 1,69) mcg par mL.
Absorption et biodisponibilité
La biodisponibilité absolue chez 12 sujets adultes était de 86 % ± 16 % (moyenne ± ET) pour le comprimé à 150 mg et de 87 % ± 13 % pour la solution buvable. Après administration orale de 2 mg par kg deux fois par jour à 9 adultes infectés par le VIH-1, la concentration sérique maximale de lamivudine (Cmax) était de 1,5 ± 0,5 mcg par mL (moyenne ± ET). L'aire sous la courbe de la concentration plasmatique en fonction du temps (ASC) et la Cmax ont augmenté proportionnellement à la dose orale sur la plage de 0,25 à 10 mg par kg.
Le rapport d'accumulation de la lamivudine chez les adultes asymptomatiques séropositifs pour le VIH-1 ayant une fonction rénale normale était de 1,50 après 15 jours d'administration orale de 2 mg par kg deux fois par jour.
Effets des aliments sur l'absorption orale
EPIVIR 150mg comprimés et solution buvable peut être administré avec ou sans nourriture. Une forme posologique expérimentale de 25 mg de lamivudine a été administrée par voie orale à 12 sujets asymptomatiques infectés par le VIH-1 à 2 reprises, une fois à jeun et une fois avec de la nourriture (1 099 kcal ; 75 grammes de matières grasses, 34 grammes de protéines, 72 grammes de glucides ). L'absorption de la lamivudine était plus lente à jeun (Tmax : 3,2 ± 1,3 heures) qu'à jeun (Tmax : 0,9 ± 0,3 heures) ; La Cmax à jeun était inférieure de 40 % ± 23 % (moyenne ± ET) à celle à jeun. Il n'y avait pas de différence significative dans l'exposition systémique (AUC∞) à jeun et à jeun.
Distribution
Le volume de distribution apparent après administration IV de lamivudine à 20 sujets était de 1,3 ± 0,4 L par kg, suggérant que la lamivudine se distribue dans les espaces extravasculaires. Le volume de distribution était indépendant de la dose et n'était pas corrélé au poids corporel.
La liaison de la lamivudine aux protéines plasmatiques humaines est inférieure à 36 %. Des études in vitro ont montré que sur la plage de concentrations de 0,1 à 100 mcg par mL, la quantité de lamivudine associée aux érythrocytes variait de 53 % à 57 % et était indépendante de la concentration.
Métabolisme
Le métabolisme de la lamivudine est une voie d'élimination mineure. Chez l'homme, le seul métabolite connu de la lamivudine est le métabolite trans-sulfoxyde (environ 5 % d'une dose orale après 12 heures). Les concentrations sériques de ce métabolite n'ont pas été déterminées. La lamivudine n'est pas significativement métabolisée par les enzymes du cytochrome P450.
Élimination
La majorité de la lamivudine est éliminée sous forme inchangée dans les urines par sécrétion cationique organique active. Chez 9 sujets sains ayant reçu une dose orale unique de 300 mg de lamivudine, la clairance rénale était de 199,7 ± 56,9 mL par minute (moyenne ± ET). Chez 20 sujets infectés par le VIH-1 ayant reçu une dose IV unique, la clairance rénale était de 280,4 ± 75,2 mL par min (moyenne ± ET), représentant 71 % ± 16 % (moyenne ± ET) de la clairance totale de la lamivudine.
Dans la plupart des essais à dose unique chez des sujets infectés par le VIH-1, des sujets infectés par le VHB ou des sujets sains avec prélèvement de sérum pendant 24 heures après l'administration, la demi-vie d'élimination moyenne observée (t½) variait de 5 à 7 heures. Chez les sujets infectés par le VIH-1, la clairance totale était de 398,5 ± 69,1 mL par min (moyenne ± ET). La clairance orale et la demi-vie d'élimination étaient indépendantes de la dose et du poids corporel sur une plage de doses orales de 0,25 à 10 mg par kg.
Populations spécifiques
Patients atteints d'insuffisance rénale
Les propriétés pharmacocinétiques de la lamivudine ont été déterminées dans un petit groupe d'adultes infectés par le VIH-1 présentant une insuffisance rénale (tableau 7).
Le Tmax n'a pas été significativement affecté par la fonction rénale. Sur la base de ces observations, il est recommandé de modifier la posologie de la lamivudine chez les insuffisants rénaux [voir DOSAGE ET ADMINISTRATION ].
Sur la base d'un essai chez des sujets par ailleurs en bonne santé présentant une fonction rénale altérée, l'hémodialyse a augmenté la clairance de la lamivudine d'une moyenne de 64 à 88 ml par minute ; cependant, la durée de l'hémodialyse (4 heures) était insuffisante pour modifier significativement l'exposition moyenne à la lamivudine après l'administration d'une dose unique. La dialyse péritonéale continue ambulatoire et la dialyse péritonéale automatisée ont des effets négligeables sur la clairance de la lamivudine. Par conséquent, il est recommandé, après correction de la dose pour la clairance de la créatinine, qu'aucune modification supplémentaire de la dose ne soit effectuée après une hémodialyse de routine ou une dialyse péritonéale.
Les effets de l'insuffisance rénale sur la pharmacocinétique de la lamivudine chez les patients pédiatriques ne sont pas connus.
Patients atteints d'insuffisance hépatique
Les propriétés pharmacocinétiques de la lamivudine ont été déterminées chez des adultes présentant une insuffisance hépatique. Les paramètres pharmacocinétiques n'ont pas été modifiés par la diminution de la fonction hépatique. L'innocuité et l'efficacité de la lamivudine n'ont pas été établies en présence d'une maladie hépatique décompensée.
Femmes enceintes
La pharmacocinétique de la lamivudine a été étudiée chez 36 femmes enceintes au cours de 2 essais cliniques menés en Afrique du Sud. La pharmacocinétique de la lamivudine chez les femmes enceintes était similaire à celle observée chez les adultes non enceintes et chez les femmes post-partum. Les concentrations de lamivudine étaient généralement similaires dans les échantillons de sérum maternel, néonatal et de cordon ombilical.
Patients pédiatriques
La pharmacocinétique de la lamivudine a été étudiée après des doses uniques ou répétées d'EPIVIR chez 210 sujets pédiatriques. Les sujets pédiatriques recevant la solution buvable de lamivudine (dosée à environ 8 mg par kg par jour) ont atteint des concentrations plasmatiques de lamivudine inférieures d'environ 25 % par rapport aux adultes infectés par le VIH-1. Les sujets pédiatriques recevant des comprimés oraux de lamivudine ont atteint des concentrations plasmatiques comparables ou légèrement supérieures à celles observées chez les adultes. La biodisponibilité absolue des comprimés EPIVIR et de la solution buvable est plus faible chez les enfants que chez les adultes. La biodisponibilité relative d'EPIVIR solution buvable est inférieure d'environ 40 % à celle des comprimés contenant de la lamivudine chez les sujets pédiatriques malgré l'absence de différence chez les adultes. Des expositions plus faibles à la lamivudine chez les patients pédiatriques recevant EPIVIR 150 mg solution buvable sont probablement dues à l'interaction entre la lamivudine et les solutions concomitantes contenant du sorbitol (telles que ZIAGEN). La modélisation des données pharmacocinétiques suggère qu'il est nécessaire d'augmenter la posologie d'EPIVIR solution buvable à 5 mg par kg par voie orale deux fois par jour ou 10 mg par kg par voie orale une fois par jour (jusqu'à un maximum de 300 mg par jour) pour atteindre des concentrations suffisantes de lamivudine [voir DOSAGE ET ADMINISTRATION ]. Il n'y a pas de données cliniques chez les patients pédiatriques infectés par le VIH-1 co-administrés avec des médicaments contenant du sorbitol à cette dose.
La pharmacocinétique de la lamivudine administrée une fois par jour chez des sujets pédiatriques infectés par le VIH-1 âgés de 3 mois à 12 ans a été évaluée dans 3 essais (PENTA-15 [n = 17], PENTA 13 [n = 19] et ARROW PK [n = 35]). Les 3 essais étaient des essais pharmacocinétiques ouverts croisés à 2 périodes portant sur l'administration d'abacavir et de lamivudine deux fois par rapport à une dose une fois par jour. Ces 3 essais ont démontré qu'une administration uniquotidienne fournit une ASC0-24 similaire à une administration biquotidienne de lamivudine à la même dose quotidienne totale lorsque l'on compare les schémas posologiques au sein de la même formulation (c'est-à-dire la solution orale ou la formulation en comprimés). La Cmax moyenne était environ 80 % à 90 % plus élevée avec la lamivudine administrée une fois par jour par rapport à l'administration biquotidienne.
La distribution de la lamivudine dans le liquide céphalo-rachidien (LCR) a été évaluée chez 38 sujets pédiatriques après plusieurs doses orales de lamivudine. Des échantillons de LCR ont été prélevés entre 2 et 4 heures après l'administration. À la dose de 8 mg par kg par jour, les concentrations de lamivudine dans le LCR chez 8 sujets variaient de 5,6 % à 30,9 % (moyenne ± ET de 14,2 % ± 7,9 %) de la concentration dans un échantillon de sérum simultané, avec des concentrations de lamivudine dans le LCR allant de 0,04 à 0,3 mcg par mL.
Des données pharmacocinétiques et de sécurité limitées et non contrôlées sont disponibles sur l'administration de lamivudine (et de zidovudine) à 36 nourrissons âgés de moins d'une semaine dans 2 essais en Afrique du Sud. Dans ces essais, la clairance de la lamivudine a été considérablement réduite chez les nouveau-nés d'une semaine par rapport aux sujets pédiatriques (âgés de plus de 3 mois) étudiés précédemment. Il n'y a pas suffisamment d'informations pour établir l'évolution dans le temps des changements de clairance entre la période néonatale immédiate et les tranches d'âge supérieures à 3 mois [voir EFFETS INDÉSIRABLES ].
Patients gériatriques
La pharmacocinétique de la lamivudine après administration d'EPIVIR 150 mg à des sujets de plus de 65 ans n'a pas été étudiée [voir Utilisation dans des populations spécifiques ].
Patients masculins et féminins
Il n'y a pas de différences significatives ou cliniquement pertinentes entre les sexes dans la pharmacocinétique de la lamivudine.
Groupes raciaux
Il n'y a pas de différences raciales significatives ou cliniquement pertinentes dans la pharmacocinétique de la lamivudine.
Études sur les interactions médicamenteuses
Effet de la lamivudine sur la pharmacocinétique d'autres agents
D'après les résultats d'études in vitro, la lamivudine à des expositions thérapeutiques aux médicaments ne devrait pas affecter la pharmacocinétique des médicaments qui sont des substrats des transporteurs suivants : polypeptide transporteur d'anions organiques 1B1/3 (OATP1B1/3), protéine de résistance au cancer du sein (BCRP), Glycoprotéine P (P-gp), protéine d'extrusion multidrogue et toxine 1 (MATE)1, MATE2-K, transporteur de cations organiques 1 (OCT)1, OCT2 ou OCT3.
Effet d'autres agents sur la pharmacocinétique de la lamivudine
La lamivudine est un substrat de MATE1, MATE2-K et OCT2 in vitro. Il a été démontré que le triméthoprime (un inhibiteur de ces transporteurs de médicaments) augmente les concentrations plasmatiques de lamivudine. Cette interaction n'est pas considérée comme cliniquement significative car aucun ajustement posologique de la lamivudine n'est nécessaire.
La lamivudine est un substrat de la P-gp et de la BCRP ; cependant, compte tenu de sa biodisponibilité absolue (87 %), il est peu probable que ces transporteurs jouent un rôle significatif dans l'absorption de la lamivudine. Par conséquent, il est peu probable que l'administration concomitante de médicaments qui sont des inhibiteurs de ces transporteurs d'efflux affecte la disposition et l'élimination de la lamivudine.
Interféron Alfa
Il n'y a pas eu d'interaction pharmacocinétique significative entre la lamivudine et l'interféron alfa dans un essai portant sur 19 hommes sains [voir AVERTISSEMENTS ET PRECAUTIONS ].
Ribavirine
Les données in vitro indiquent que la ribavirine réduit la phosphorylation de la lamivudine, de la stavudine et de la zidovudine. Cependant, aucune interaction pharmacocinétique (p. ex., concentrations plasmatiques ou concentrations intracellulaires de métabolites actifs triphosphorylés) ou pharmacodynamique (p. ex., perte de la suppression virologique du VIH-1/VHC) n'a été observée lorsque la ribavirine et la lamivudine (n = 18), la stavudine (n = 10) , ou de la zidovudine (n = 6) ont été co-administrés dans le cadre d'un traitement multimédicamenteux à des sujets co-infectés par le VIH-1/VHC [voir AVERTISSEMENTS ET PRECAUTIONS ].
Sorbitol (Excipient)
Des solutions de lamivudine et de sorbitol ont été co-administrées à 16 sujets adultes sains dans un essai croisé ouvert, à séquence aléatoire, à 4 périodes. Chaque sujet a reçu une dose unique de 300 mg de solution buvable de lamivudine seule ou co-administrée avec une dose unique de 3,2 grammes, 10,2 grammes ou 13,4 grammes de sorbitol en solution. L'administration concomitante de lamivudine et de sorbitol a entraîné des diminutions dose-dépendantes de 20 %, 39 % et 44 % de l'ASC(0-24), 14 %, 32 % et 36 % de l'ASC(∞) et de 28 %, 52 % et 55 % dans la Cmax ; de lamivudine, respectivement.
Triméthoprime/Sulfaméthoxazole
La lamivudine et le TMP/SMX ont été co-administrés à 14 sujets séropositifs pour le VIH-1 dans le cadre d'un essai croisé monocentrique, ouvert, randomisé. Chaque sujet a reçu un traitement avec une dose unique de 300 mg de lamivudine et de TMP 160 mg/SMX 800 mg une fois par jour pendant 5 jours avec une administration concomitante de lamivudine 300 mg avec la cinquième dose dans un schéma croisé. L'administration concomitante de TMP/SMX avec la lamivudine a entraîné une augmentation de 43 % ± 23 % (moyenne ± ET) de l'ASC∞ de la lamivudine, une diminution de 29 % ± 13 % de la clairance orale de la lamivudine et une diminution de 30 % ± 36 % de la clairance rénale de la lamivudine. Les propriétés pharmacocinétiques du TMP et du SMX n'ont pas été modifiées par la co-administration avec la lamivudine. Il n'existe aucune information concernant l'effet sur la pharmacocinétique de la lamivudine de doses plus élevées de TMP/SMX telles que celles utilisées dans le traitement du PCP.
Zidovudine
Aucune altération cliniquement significative de la pharmacocinétique de la lamivudine ou de la zidovudine n'a été observée chez 12 sujets asymptomatiques infectés par le VIH-1 ayant reçu une dose unique de zidovudine (200 mg) en association avec des doses multiples de lamivudine (300 mg toutes les 12 heures).
Microbiologie
Mécanisme d'action
La lamivudine est un analogue nucléosidique synthétique. Au niveau intracellulaire, la lamivudine est phosphorylée en son métabolite actif 5'-triphosphate, la lamivudine triphosphate (3TC-TP). Le principal mode d'action du 3TC-TP est l'inhibition de la transcriptase inverse (RT) du VIH-1 via la terminaison de la chaîne d'ADN après incorporation de l'analogue nucléotidique.
Activité antivirale
L'activité antivirale de la lamivudine contre le VIH-1 a été évaluée dans un certain nombre de lignées cellulaires, y compris les monocytes et les lymphocytes du sang périphérique humain frais (PBMC) à l'aide de tests de sensibilité standard. Les valeurs d'EC50 étaient comprises entre 0,003 et 15 microM (1 microM = 0,23 mcg par mL). Les valeurs médianes d'EC50 de la lamivudine étaient de 60 nM (intervalle : 20 à 70 nM), 35 nM (intervalle : 30 à 40 nM), 30 nM (intervalle : 20 à 90 nM), 20 nM (intervalle : 3 à 40 nM) , 30 nM (plage : 1 à 60 nM), 30 nM (plage : 20 à 70 nM), 30 nM (plage : 3 à 70 nM) et 30 nM (plage : 20 à 90 nM) contre les clades du VIH-1 Virus AG et groupe O (n = 3 sauf n = 2 pour le clade B) respectivement. Les valeurs de CE50 contre les isolats du VIH-2 (n = 4) variaient de 0,003 à 0,120 microM dans les PBMC. La lamivudine n'était pas antagoniste de tous les agents anti-VIH testés. La ribavirine (50 μM) utilisée dans le traitement de l'infection chronique par le VHC a diminué l'activité anti-VIH-1 de la lamivudine de 3,5 fois dans les cellules MT-4.
La résistance
Des variants du VIH-1 résistants à la lamivudine ont été sélectionnés en culture cellulaire. L'analyse génotypique a montré que la résistance était due à une substitution spécifique d'acides aminés dans la transcriptase inverse du VIH-1 au codon 184, changeant la méthionine en valine ou en isoleucine (M184V/I).
Des souches de VIH-1 résistantes à la fois à la lamivudine et à la zidovudine ont été isolées chez des sujets. La sensibilité des isolats cliniques à la lamivudine et à la zidovudine a été surveillée dans des essais cliniques contrôlés. Chez les sujets recevant la lamivudine en monothérapie ou en association avec la lamivudine et la zidovudine, les isolats de VIH-1 de la plupart des sujets sont devenus phénotypiquement et génotypiquement résistants à la lamivudine dans les 12 semaines.
Analyse génotypique et phénotypique des isolats du VIH-1 sous traitement provenant de sujets présentant un échec virologique
Essai EPV20001
Cinquante-trois des 554 (10 %) sujets inscrits à EPV20001 ont été identifiés comme des échecs virologiques (taux plasmatique d'ARN du VIH-1 supérieur ou égal à 400 copies par ml) à la semaine 48. Vingt-huit sujets ont été randomisés pour recevoir la lamivudine une fois groupe de traitement quotidien et 25 au groupe de traitement lamivudine deux fois par jour. Les taux plasmatiques initiaux médians d'ARN du VIH-1 des sujets du groupe lamivudine une fois par jour et du groupe lamivudine deux fois par jour étaient de 4,9 log10 copies par ml et de 4,6 log10 copies par ml, respectivement.
L'analyse génotypique des isolats sous traitement de 22 sujets identifiés comme des échecs virologiques dans le groupe lamivudine une fois par jour a montré que les isolats de 8 des 22 sujets contenaient une substitution associée à la résistance à la lamivudine apparue sous traitement (M184V ou M184I), les isolats de 0 sur 22 les sujets contenaient des substitutions d'acides aminés apparues sous traitement associées à la résistance à la zidovudine (M41L, D67N, K70R, L210W, T215Y/F ou K219Q/E), et les isolats de 10 des 22 sujets contenaient des substitutions d'acides aminés apparues sous traitement associées à la résistance à l'éfavirenz ( L100I, K101E, K103N, V108I ou Y181C).
L'analyse génotypique des isolats sous traitement des sujets (n = 22) du groupe de traitement par la lamivudine deux fois par jour a montré que les isolats de 5 des 22 sujets contenaient des substitutions de résistance à la lamivudine apparues sous traitement, les isolats de 1 des 22 sujets contenaient une résistance à la zidovudine apparue sous traitement substitutions, et les isolats de 7 des 22 sujets contenaient des substitutions de résistance à l'éfavirenz apparues sous traitement.
L'analyse phénotypique d'isolats de VIH-1 appariés à l'inclusion sous traitement chez des sujets (n = 13) recevant de la lamivudine une fois par jour a montré que les isolats de 7 des 13 sujets présentaient une diminution de 85 à 299 fois de la sensibilité à la lamivudine, les isolats de 12 des 13 sujets étaient sensibles à la zidovudine, et les isolats de 8 des 13 sujets présentaient une diminution de 25 à 295 fois de la sensibilité à l'éfavirenz.
L'analyse phénotypique d'isolats de VIH-1 appariés au départ sous traitement provenant de sujets (n = 13) recevant de la lamivudine deux fois par jour a montré que les isolats de 4 des 13 sujets présentaient une diminution de 29 à 159 fois de la sensibilité à la lamivudine, les isolats des 13 les sujets étaient sensibles à la zidovudine et les isolats de 3 des 13 sujets présentaient une diminution de 21 à 342 fois de la sensibilité à l'éfavirenz.
Essai EPV40001
Cinquante sujets ont reçu 300 mg de lamivudine une fois par jour plus 300 mg de zidovudine deux fois par jour plus 300 mg d'abacavir deux fois par jour et 50 sujets ont reçu 150 mg de lamivudine plus 300 mg de zidovudine plus 300 mg d'abacavir deux fois par jour. Les niveaux médians d'ARN du VIH-1 dans le plasma de base pour les sujets des 2 groupes étaient de 4,79 log10 copies par ml et de 4,83 log10 copies par ml, respectivement. Quatorze des 50 sujets du groupe de traitement lamivudine une fois par jour et 9 des 50 sujets du groupe lamivudine deux fois par jour ont été identifiés comme des échecs virologiques.
L'analyse génotypique des isolats de VIH-1 sous traitement provenant de sujets (n = 9) du groupe de traitement par la lamivudine une fois par jour a montré que les isolats de 6 sujets avaient une substitution M184V associée à la résistance à l'abacavir et/ou à la lamivudine seule. Les isolats sous traitement de sujets (n = 6) recevant de la lamivudine deux fois par jour ont montré que les isolats de 2 sujets avaient M184V seul, et que les isolats de 2 sujets portaient la substitution M184V en combinaison avec des substitutions d'acides aminés associées à la résistance à la zidovudine.
L'analyse phénotypique des isolats sous traitement de sujets (n = 6) recevant de la lamivudine une fois par jour a montré que les isolats de VIH-1 de 4 sujets présentaient une diminution de 32 à 53 fois de la sensibilité à la lamivudine. Les isolats de VIH-1 de ces 6 sujets étaient sensibles à la zidovudine.
L'analyse phénotypique des isolats sous traitement de sujets (n = 4) recevant de la lamivudine deux fois par jour a montré que les isolats de VIH-1 d'un sujet présentaient une diminution de 45 fois de la sensibilité à la lamivudine et une diminution de 4,5 fois de la sensibilité à la zidovudine.
Pédiatrie
Les sujets pédiatriques recevant la solution buvable de lamivudine en même temps que d'autres solutions buvables antirétrovirales (abacavir, névirapine/éfavirenz ou zidovudine) dans ARROW ont développé une résistance virale plus fréquemment que ceux recevant des comprimés. Lors de la randomisation à une dose quotidienne ou biquotidienne d'EPIVIR plus abacavir, 13 % des sujets qui ont commencé les comprimés et 32 % des sujets qui ont commencé la solution avaient des substitutions de résistance. Le profil de résistance observé en pédiatrie est similaire à celui observé chez l'adulte en termes de substitutions génotypiques détectées et de fréquence relative, avec les substitutions les plus fréquemment détectées à M184 (V ou I) [voir Etudes cliniques ].
Résistance croisée
Une résistance croisée a été observée parmi les inhibiteurs nucléosidiques de la transcriptase inverse (INTI). Les mutants du VIH-1 résistants à la lamivudine présentaient une résistance croisée en culture cellulaire à la didanosine (ddI). Une résistance croisée est également attendue avec l'abacavir et l'emtricitabine car ces substitutions sélectionnent M184V.
Etudes cliniques
L'utilisation d'EPIVIR est basée sur les résultats d'essais cliniques chez des sujets infectés par le VIH-1 dans des schémas thérapeutiques combinés avec d'autres agents antirétroviraux. Les informations provenant d'essais avec des critères d'évaluation cliniques ou une combinaison de numération des cellules CD4+ et de mesures de l'ARN du VIH-1 sont incluses ci-dessous pour documenter la contribution de la lamivudine à un traitement combiné dans des essais contrôlés.
Sujets adultes
Essai clinique sur les critères d'évaluation
NUCB3007 (CAESAR) était un essai multicentrique, en double aveugle, contrôlé par placebo comparant la poursuite du traitement actuel (zidovudine seule [62 % des sujets] ou zidovudine avec didanosine ou zalcitabine [38 % des sujets]) à l'ajout d'EPIVIR 150 mg ou d'EPIVIR 150 mg plus un inhibiteur expérimental non nucléosidique de la transcriptase inverse (INNTI), randomisé 1:2:1. Un total de 1 816 adultes infectés par le VIH-1 avec 25 à 250 cellules CD4+ par mm³ (médiane = 122 cellules par mm³) au départ ont été recrutés : l'âge médian était de 36 ans, 87 % étaient des hommes, 84 % avaient été exposés aux nucléosides et 16 % étaient naïfs de traitement. La durée médiane de l'essai était de 12 mois. Les résultats sont résumés dans le tableau 9.
Essais de critère de substitution
Essais d'analogues à double nucléoside
Les principaux essais cliniques du développement initial de la lamivudine ont comparé les associations lamivudine/zidovudine à la zidovudine en monothérapie ou à la zidovudine plus zalcitabine. Ces essais ont démontré l'effet antiviral de la lamivudine dans une combinaison de 2 médicaments. Des utilisations plus récentes de la lamivudine dans le traitement de l'infection par le VIH-1 l'intègrent dans des schémas thérapeutiques à plusieurs médicaments contenant au moins 3 médicaments antirétroviraux pour une suppression virale améliorée.
Comparaison des schémas posologiques Essais de critère d'évaluation de substitution chez des adultes naïfs de traitement
EPV20001 était un essai contrôlé multicentrique, en double aveugle, dans lequel les sujets étaient randomisés 1:1 pour recevoir EPIVIR 300 mg une fois par jour ou EPIVIR 150 mg deux fois par jour, en association avec la zidovudine 300 mg deux fois par jour et l'efavirenz 600 mg une fois par jour. Un total de 554 adultes infectés par le VIH-1 et naïfs de traitement antirétroviral inscrits : hommes (79 %), blancs (50 %), âge médian de 35 ans, numération initiale des cellules CD4+ de 69 à 1 089 cellules par mm³ (médiane = 362 cellules par mm³) et l'ARN du VIH-1 plasmatique initial médian de 4,66 log10 copies par ml. Les résultats du traitement sur 48 semaines sont résumés dans la figure 1 et le tableau 10.
Figure 1 : Réponse virologique jusqu'à la semaine 48, EPV20001a,b (intention de traiter) 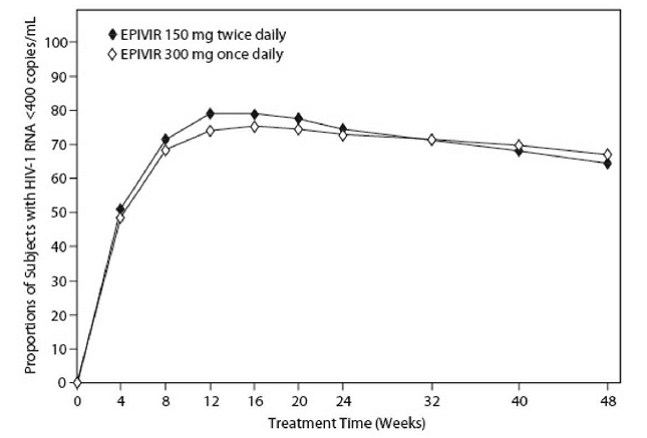
un MONITEUR Roche AMPLICOR HIV-1. b Les répondeurs à chaque visite sont des sujets qui avaient atteint et maintenu un taux d'ARN du VIH-1 inférieur à 400 copies par ml sans interruption à cette visite.
Les proportions de sujets avec un ARN du VIH-1 inférieur à 50 copies par ml (via le test Roche Ultrasensible) jusqu'à la semaine 48 étaient de 61 % pour les sujets recevant EPIVIR à 300 mg une fois par jour et de 63 % pour les sujets recevant EPIVIR à 150 mg deux fois par jour. Les augmentations médianes du nombre de cellules CD4+ étaient de 144 cellules par mm³ à la semaine 48 chez les sujets recevant EPIVIR 300 mg une fois par jour et de 146 cellules par mm³ chez les sujets recevant EPIVIR 150 mg deux fois par jour.
Un petit essai pilote randomisé et ouvert, EPV40001, a été mené en Thaïlande. Un total de 159 sujets adultes naïfs de traitement (hommes 32 %, asiatiques 100 %, âge médian 30 ans, numération initiale médiane des cellules CD4+ 380 cellules par mm³, ARN plasmatique médian du VIH-1 4,8 log10 copies par mL) ont été inscrits. Deux des bras de traitement de cet essai ont fourni une comparaison entre la lamivudine 300 mg une fois par jour (n = 54) et la lamivudine 150 mg deux fois par jour (n = 52), chacun en association avec la zidovudine 300 mg deux fois par jour et l'abacavir 300 mg deux fois par jour. Dans les analyses en intention de traiter des données sur 48 semaines, les proportions de sujets dont l'ARN du VIH-1 était inférieur à 400 copies par ml étaient de 61 % (33 sur 54) dans le groupe randomisé pour recevoir la lamivudine une fois par jour et de 75 % (39 sur 54) 52) dans le groupe randomisé pour recevoir les 3 médicaments deux fois par jour ; les proportions d'ARN du VIH-1 inférieures à 50 copies par ml étaient de 54 % (29 sur 54) dans le groupe lamivudine une fois par jour et de 67 % (35 sur 52) dans le groupe tous deux fois par jour ; et les augmentations médianes du nombre de cellules CD4+ étaient de 166 cellules par mm³ dans le groupe lamivudine une fois par jour et de 216 cellules par mm³ dans le groupe deux fois par jour.
Sujets pédiatriques
Essai clinique sur les critères d'évaluation
ACTG300 était un essai multicentrique, randomisé, en double aveugle qui prévoyait la comparaison d'EPIVIR plus RETROVIR (zidovudine) avec la didanosine en monothérapie. Un total de 471 sujets pédiatriques symptomatiques, infectés par le VIH-1 et naïfs de traitement (moins de ou égal à 56 jours de traitement antirétroviral) ont été inclus dans ces 2 bras de traitement. L'âge médian était de 2,7 ans (intervalle : 6 semaines à 14 ans), 58 % étaient des femmes et 86 % n'étaient pas de race blanche. La numération initiale moyenne des cellules CD4+ était de 868 cellules par mm³ (moyenne : 1 060 cellules par mm³ et intervalle : 0 à 4 650 cellules par mm³ pour les sujets âgés de moins de ou égal à 5 ans ; moyenne : 419 cellules par mm³ et intervalle : 0 à 1 555 cellules par mm³ pour les sujets âgés de plus de 5 ans) et l'ARN VIH-1 plasmatique initial moyen était de 5,0 log10 copies par ml. La durée médiane de l'essai était de 10,1 mois pour les sujets recevant EPIVIR 150 mg plus RETROVIR et de 9,2 mois pour les sujets recevant la didanosine en monothérapie. Les résultats sont résumés dans le tableau 11.
Dosage une fois par jour
ARROW (COL105677) était un essai multicentrique randomisé de 5 ans qui évaluait plusieurs aspects de la prise en charge clinique de l'infection par le VIH-1 chez des sujets pédiatriques. Des sujets infectés par le VIH-1 et naïfs de traitement âgés de 3 mois à 17 ans ont été recrutés et traités avec un schéma thérapeutique de première intention contenant EPIVIR 150 mg et de l'abacavir, administré deux fois par jour conformément aux recommandations de l'Organisation mondiale de la santé. Après un minimum de 36 semaines de traitement, les sujets ont eu la possibilité de participer à la randomisation 3 de l'essai ARROW, comparant l'innocuité et l'efficacité d'une administration uniquotidienne à une administration biquotidienne d'EPIVIR et d'abacavir, en association avec un troisième antirétroviral médicament, pendant 96 semaines supplémentaires. Sur les 1 206 sujets ARROW originaux, 669 ont participé à la randomisation 3. La suppression virologique n'était pas une exigence de participation : au départ de la randomisation 3 (après un minimum de 36 semaines de traitement biquotidien), 75 % des sujets du traitement biquotidien cohorte ont été virologiquement supprimées, contre 71 % des sujets de la cohorte à prise unique quotidienne.
La proportion de sujets avec un ARN du VIH-1 inférieur à 80 copies par ml pendant 96 semaines est présentée dans le tableau 12. Les différences entre les réponses virologiques dans les deux bras de traitement étaient comparables pour les caractéristiques de base pour le sexe et l'âge.
Les analyses par formulation ont démontré que la proportion de sujets ayant un ARN VIH-1 inférieur à 80 copies par ml lors de la randomisation et la semaine 96 était plus élevée chez les sujets ayant reçu des formulations de comprimés d'EPIVIR 150 mg et d'abacavir (75 % [458/610] et 72 % [434/601]) que chez ceux qui avaient reçu une ou plusieurs formulations de solution (avec une solution d'EPIVIR à 150 mg administrée à des doses basées sur la tranche de poids d'environ 8 mg par kg par jour) à tout moment (52 % [29/56] et 54 % [30/56]), respectivement [voir AVERTISSEMENTS ET PRECAUTIONS ]. Ces différences ont été observées dans chaque groupe d'âge différent évalué.
INFORMATIONS PATIENTS
ÉPIVIR (EP-i-veer) (lamivudine) comprimés
ÉPIVIR (EP-i-veer) (lamivudine) solution buvable
Quelle est l'information la plus importante que je devrais connaître sur EPIVIR 150mg ?
EPIVIR peut provoquer des effets secondaires graves, notamment :
- Aggravation du virus de l'hépatite B chez les personnes infectées par le VIH-1. Si vous êtes infecté par le VIH-1 (virus de l'immunodéficience humaine de type 1) et le virus de l'hépatite B (VHB), votre VHB peut s'aggraver (poussée) si vous arrêtez de prendre EPIVIR. Une « poussée » se produit lorsque votre infection par le VHB revient soudainement d'une manière pire qu'avant. L'aggravation de la maladie du foie peut être grave et entraîner la mort.
- Ne manquez pas d'EPIVIR. Rechargez votre ordonnance ou parlez à votre fournisseur de soins de santé avant que votre EPIVIR ne soit épuisé.
- N'arrêtez pas EPIVIR sans parler d'abord à votre pourvoyeur de soins médicaux.
- Si vous arrêtez de prendre EPIVIR, votre fournisseur de soins de santé devra vérifier votre état de santé souvent et faire des analyses de sang régulièrement pendant plusieurs mois pour vérifier votre foie.
- Virus résistant de l'hépatite B (VHB). Si vous avez le VIH-1 et l'hépatite B, le virus de l'hépatite B peut changer (muter) pendant votre traitement par EPIVIR 150 mg et devenir plus difficile à traiter (résistant).
- Utiliser avec des régimes à base d'interféron et de ribavirine. Une aggravation d'une maladie du foie ayant entraîné la mort s'est produite chez des personnes infectées à la fois par le VIH-1 et le virus de l'hépatite C qui prennent des médicaments antirétroviraux et qui sont également traitées pour l'hépatite C avec de l'interféron avec ou sans ribavirine. Si vous prenez EPIVIR 150 mg et l'interféron avec ou sans ribavirine, informez votre professionnel de la santé si vous présentez de nouveaux symptômes.
Qu'est-ce qu'EPIVIR ?
EPIVIR est un médicament délivré sur ordonnance utilisé avec d'autres médicaments antirétroviraux pour traiter l'infection par le virus de l'immunodéficience humaine (VIH-1).
Le VIH-1 est le virus responsable du syndrome d'immunodéficience acquise (SIDA).
Les comprimés et la solution buvable EPIVIR (utilisés pour traiter l'infection par le VIH-1) contiennent une dose plus élevée du même principe actif (lamivudine) que celle contenue dans le médicament EPIVIR-HBV, comprimés et solution buvable (utilisés pour traiter le VHB). Si vous avez à la fois le VIH-1 et le VHB, vous ne devez pas utiliser EPIVIR-VHB pour traiter vos infections.
La sécurité et l'efficacité d'EPIVIR n'ont pas été établies chez les enfants de moins de 3 mois.
Qui ne devrait pas prendre EPIVIR 150mg ?
Ne prenez jamais EPIVIR si vous êtes allergique à la lamivudine ou à l'un des ingrédients d'EPIVIR. Voir la fin de cette brochure d'Information Patiente pour une liste complète d'ingrédients dans EPIVIR.
Que dois-je dire à mon fournisseur de soins de santé avant de prendre EPIVIR ?
Avant de prendre EPIVIR 150 mg, informez votre professionnel de la santé si vous :
- avez ou avez eu des problèmes de foie, y compris une infection par le virus de l'hépatite B ou C.
- avoir des problèmes rénaux.
- avoir le diabète. Chaque dose de 15 mL (150 mg) de solution buvable EPIVIR contient 3 grammes de saccharose.
- êtes enceinte ou envisagez de devenir enceinte. La prise d'EPIVIR pendant la grossesse n'a pas été associée à un risque accru de malformations congénitales. Parlez à votre fournisseur de soins de santé si vous êtes enceinte ou prévoyez de devenir enceinte. Registre des grossesses. Il existe un registre des grossesses pour les femmes qui prennent des médicaments antirétroviraux pendant la grossesse. Le but de ce registre est de recueillir des informations sur votre santé et celle de votre bébé. Discutez avec votre fournisseur de soins de santé de la façon dont vous pouvez participer à ce registre.
- allaitez ou envisagez d'allaiter. N'allaitez pas si vous prenez EPIVIR.
- Vous ne devez pas allaiter si vous avez le VIH-1 en raison du risque de transmission du VIH-1 à votre bébé.
Informez votre professionnel de la santé de tous les médicaments que vous prenez, y compris les médicaments sur ordonnance et en vente libre, les vitamines et les suppléments à base de plantes.
Certains médicaments interagissent avec EPIVIR. Gardez une liste de vos médicaments et montrez-la à votre fournisseur de soins de santé et à votre pharmacien lorsque vous recevez un nouveau médicament. Vous pouvez demander à votre professionnel de la santé ou à votre pharmacien une liste des médicaments qui interagissent avec EPIVIR.
Ne commencez pas à prendre un nouveau médicament sans en parler à votre fournisseur de soins de santé. Votre fournisseur de soins de santé peut vous dire s'il est sécuritaire de prendre EPIVIR avec d'autres médicaments.
Comment dois-je prendre EPIVIR ?
- Prenez EPIVIR exactement comme votre fournisseur de soins de santé vous dit de le prendre.
- Si vous manquez une dose d'EPIVIR 150 mg, prenez-la dès que vous vous en souvenez. Ne prenez pas 2 doses en même temps ou ne prenez pas plus que ce que votre professionnel de la santé vous a dit de prendre.
- Restez sous la garde d'un fournisseur de soins de santé pendant le traitement avec EPIVIR.
- EPIVIR peut être pris avec ou sans nourriture.
- Pour les enfants de 3 mois et plus, votre fournisseur de soins de santé prescrira une dose d'EPIVIR 150 mg en fonction du poids corporel de votre enfant.
- Informez votre fournisseur de soins de santé si vous ou votre enfant avez de la difficulté à avaler des comprimés. EPIVIR 150mg se présente également sous forme liquide (solution buvable).
- Ne manquez pas d'EPIVIR. Le virus dans votre sang peut augmenter et le virus peut devenir plus difficile à traiter. Lorsque votre approvisionnement commence à s'épuiser, obtenez-en davantage auprès de votre fournisseur de soins de santé ou de votre pharmacie.
- Si vous avez pris trop d'EPIVIR 150 mg, appelez votre professionnel de la santé ou rendez-vous immédiatement aux urgences de l'hôpital le plus proche.
Quels sont les effets secondaires possibles d'EPIVIR 150mg ?
- EPIVIR 150 mg peut provoquer des effets secondaires graves, notamment :
- Voir « Quelles sont les informations les plus importantes que je devrais connaître sur EPIVIR 150 mg ? »
- Accumulation d'un acide dans votre sang (acidose lactique). L'acidose lactique peut survenir chez certaines personnes qui prennent EPIVIR. L'acidose lactique est une urgence médicale grave qui peut entraîner la mort. Appelez immédiatement votre fournisseur de soins de santé si vous présentez l'un des symptômes suivants qui pourraient être des signes d'acidose lactique :
- se sentir très faible ou fatigué
- avoir froid, surtout dans les bras et les jambes
- douleur musculaire inhabituelle (pas normale)
- se sentir étourdi ou étourdi
- difficulté à respirer
- avoir un rythme cardiaque rapide ou irrégulier
- douleurs à l'estomac avec nausées et vomissements
- Graves problèmes de foie peut se produire chez les personnes qui prennent EPIVIR. Dans certains cas, ces problèmes hépatiques graves peuvent entraîner la mort. Votre foie peut grossir (hépatomégalie) et vous pouvez développer de la graisse dans votre foie (stéatose). Appelez immédiatement votre fournisseur de soins de santé si vous présentez l'un des signes ou symptômes suivants de problèmes de foie :
- votre peau ou la partie blanche de vos yeux devient jaune (jaunisse)
- perte d'appétit pendant plusieurs jours ou plus
- nausée
- urine foncée ou « couleur thé »
- douleur, courbatures ou sensibilité du côté droit de la région de votre estomac
- selles de couleur claire (selles)
Vous pourriez être plus susceptible de développer une acidose lactique ou de graves problèmes de foie si vous êtes une femme ou si vous êtes en surpoids (obèse).
- Risque d'inflammation du pancréas (pancréatite). Les enfants peuvent être à risque de développer une pancréatite pendant le traitement par EPIVIR s'ils :
- avez pris des médicaments analogues nucléosidiques
- avez des antécédents de pancréatite dans le passé
- ont d'autres facteurs de risque de pancréatite
Appelez immédiatement votre fournisseur de soins de santé si votre enfant présente des signes et des symptômes de pancréatite, notamment des douleurs intenses dans la partie supérieure de l'estomac, avec ou sans nausées et vomissements. Votre professionnel de la santé peut vous dire d'arrêter de donner EPIVIR 150 mg à votre enfant si ses symptômes et les résultats des tests sanguins montrent que votre enfant peut avoir une pancréatite.
- Modifications de votre système immunitaire (syndrome de reconstitution immunitaire) peut survenir lorsque vous commencez à prendre des médicaments contre le VIH-1. Votre système immunitaire peut se renforcer et commencer à combattre des infections cachées depuis longtemps dans votre corps. Informez immédiatement votre fournisseur de soins de santé si vous commencez à avoir de nouveaux symptômes après avoir commencé à prendre EPIVIR.
Les effets indésirables les plus fréquents d'EPIVIR 150 mg chez l'adulte incluent :
- mal de tête
- signes et symptômes nasaux
- nausée
- diarrhée
- ne se sent généralement pas bien
- fatigue
- toux
Les effets indésirables les plus fréquents d'EPIVIR 150 mg chez l'enfant sont la fièvre et la toux.
Dites à votre fournisseur de soins de santé si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles d'EPIVIR. Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
Comment dois-je conserver EPIVIR ?
- Conservez les comprimés EPIVIR 150mg et la solution buvable à température ambiante entre 68°F et 77°F (20°C et 25°C).
- Conserver les flacons d'EPIVIR 150mg solution buvable bien fermés.
Gardez EPIVIR et tous les médicaments hors de la portée des enfants.
Informations générales sur l'utilisation sûre et efficace d'EPIVIR.
Les médicaments sont parfois prescrits à des fins autres que celles énumérées dans une notice d'information destinée aux patients. N'utilisez pas EPIVIR pour une affection pour laquelle il n'a pas été prescrit. Ne donnez pas EPIVIR à d'autres personnes, même si elles présentent les mêmes symptômes que vous. Cela peut leur nuire.
Vous pouvez demander à votre professionnel de la santé ou à votre pharmacien des informations sur EPIVIR 150 mg destinées aux professionnels de la santé.
Pour plus d'informations, rendez-vous sur www.viivhealthcare.com ou appelez le 1-877-844-8872.
Quels sont les ingrédients d'EPIVIR 150mg ?
Ingrédient actif : lamivudine
Ingrédients inactifs:
EPIVIR 150 mg, comprimés pelliculés sécables de 150 mg : hypromellose, stéarate de magnésium, cellulose microcristalline, polyéthylène glycol, polysorbate 80, glycolate d'amidon sodique et dioxyde de titane.
EPIVIR 300 mg comprimés pelliculés : oxyde de fer noir, hypromellose, stéarate de magnésium, cellulose microcristalline, polyéthylène glycol, polysorbate 80, glycolate d'amidon sodique et dioxyde de titane.
EPIVIR 150mg solution buvable : arômes artificiels fraise et banane, acide citrique (anhydre), méthylparabène, propylène glycol, propylparabène, citrate de sodium (dihydraté), et saccharose (200 mg par mL).
Ces informations destinées aux patients ont été approuvées par la Food and Drug Administration des États-Unis.