Rebetol 200mg Ribavirin Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce que Rebetol 200 mg et comment est-il utilisé ?
Rebetol est un médicament délivré sur ordonnance utilisé pour traiter les symptômes de l'hépatite C chronique. Rebetol 200 mg peut être utilisé seul ou avec d'autres médicaments.
Rebetol appartient à une classe de médicaments appelés agents de l'hépatite B/hépatite C ; Agents VRS.
On ne sait pas si Rebetol 200 mg est sûr et efficace chez les enfants de moins de 3 ans.
Quels sont les effets secondaires possibles de Rebetol ?
Rebetol peut provoquer des effets secondaires graves, notamment :
- urticaire,
- difficulté à respirer,
- gonflement du visage, des lèvres, de la langue ou de la gorge,
- problèmes de vue
- douleur intense dans le haut de l'estomac se propageant à votre dos,
- nausée,
- vomissement,
- diarrhée,
- toux nouvelle ou qui s'aggrave,
- fièvre,
- douleur thoracique lancinante,
- respiration sifflante,
- essoufflement,
- dépression sévère,
- pensées d'automutilation,
- pensées de blesser les autres,
- peau pâle ou jaunie,
- urine de couleur foncée,
- confusion,
- la faiblesse,
- des frissons,
- symptômes pseudo-grippaux,
- gencives enflées,
- plaies buccales,
- plaies cutanées,
- ecchymoses faciles,
- saignements inhabituels et
- étourdissement
- Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
- Les effets secondaires les plus courants de Rebetol incluent :
- nausée,
- vomissement,
- perte d'appétit,
- fièvre,
- des frissons,
- tremblement,
- faible nombre de cellules sanguines,
- anémie,
- la faiblesse,
- fatigue,
- mal de tête,
- douleur musculaire,
- des changements d'humeur,
- l'anxiété, et
- irritabilité
Dites au médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles de Rebetol. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
ATTENTION
RISQUE DE TROUBLES GRAVES ET EFFETS ASSOCIÉS À LA RIBAVIRINE
- REBETOL en monothérapie n'est pas efficace pour le traitement de l'infection chronique par le virus de l'hépatite C et ne doit pas être utilisé seul pour cette indication [voir MISES EN GARDE ET PRÉCAUTIONS].
- La principale toxicité de la ribavirine est l'anémie hémolytique. L'anémie associée au traitement par REBETOL 200 mg peut entraîner une aggravation de la maladie cardiaque qui a conduit à des infarctus du myocarde mortels et non mortels. Les patients ayant des antécédents de maladie cardiaque importante ou instable ne doivent pas être traités par REBETOL [voir POSOLOGIE ET ADMINISTRATION, MISES EN GARDE ET PRÉCAUTIONS et RÉACTIONS INDÉSIRABLES].
- Des effets tératogènes et embryocides significatifs ont été démontrés chez toutes les espèces animales exposées à la ribavirine. De plus, la ribavirine a une demi-vie à doses multiples de 12 jours, et elle peut donc persister dans les compartiments non plasmatiques jusqu'à 6 mois. Par conséquent, le traitement par REBETOL est contre-indiqué chez les femmes enceintes et chez les partenaires masculins des femmes enceintes. Le soin extrême doit être pris pour éviter la grossesse pendant la thérapie et depuis 6 mois après l'achèvement de traitement tant dans les patients féminins que dans les partenaires féminins de patients masculins qui prennent la thérapie REBETOL. Au moins deux formes fiables de contraception efficace doivent être utilisées pendant le traitement et pendant la période de suivi post-traitement de 6 mois [voir CONTRE-INDICATIONS, MISES EN GARDE ET PRÉCAUTIONS, Utilisation dans des populations spécifiques, Toxicologie non clinique et RENSEIGNEMENTS POUR LES PATIENTS].
LA DESCRIPTION
REBETOL (ribavirine), est un analogue nucléosidique synthétique (analogue purique). Le nom chimique de la ribavirine est 1-β-D-ribofuranosyl-1H-1,2,4-triazole-3-carboxamide et a la formule développée suivante (voir Figure 1).
Figure 1 : Formule structurelle
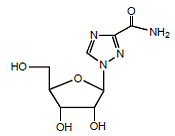
La ribavirine est une poudre cristalline blanche. Il est librement soluble dans l'eau et légèrement soluble dans l'alcool anhydre. La formule empirique est C8H12N4O5 et le poids moléculaire est de 244,21.
Les capsules REBETOL consistent en une poudre blanche dans une capsule de gélatine blanche et opaque. Chaque gélule contient 200 mg de ribavirine et les ingrédients inactifs suivants : cellulose microcristalline, lactose monohydraté, croscarmellose sodique et stéarate de magnésium. L'enveloppe de la gélule est constituée de gélatine, de laurylsulfate de sodium, de dioxyde de silicium et de dioxyde de titane. La capsule est imprimée avec de l'encre pharmaceutique bleue comestible composée de gomme laque, d'alcool éthylique anhydre, d'alcool isopropylique, d'alcool n-butylique, de propylène glycol, d'hydroxyde d'ammonium et de laque d'aluminium FD&C Blue #2.
REBETOL 200mg solution buvable est un liquide clair, incolore à pâle ou jaune clair aromatisé au chewing-gum. Chaque millilitre de la solution contient 40 mg de ribavirine et les ingrédients inactifs saccharose, glycérine, sorbitol, propylène glycol, citrate de sodium, acide citrique, benzoate de sodium, arôme naturel et artificiel de chewing-gum #15864 et eau.
LES INDICATIONS
Hépatite C chronique (HCC)
REBETOL® (ribavirine) en association avec l'interféron alfa-2b (pégylé et non pégylé) est indiqué pour le traitement de l'hépatite C chronique (HCC) chez les patients de 3 ans et plus atteints d'une maladie hépatique compensée [voir AVERTISSEMENTS ET PRECAUTIONS , et Utilisation dans des populations spécifiques ].
Les points suivants doivent être pris en compte lors de l'instauration d'un traitement combiné REBETOL avec PegIntron® ou INTRON A® :
- Ces indications sont basées sur l'obtention d'un ARN-VHC indétectable après un traitement de 24 ou 48 semaines et le maintien d'une réponse virologique soutenue (RVS) 24 semaines après la dernière dose.
- La thérapie combinée avec REBETOL/PegIntron est préférée à REBETOL/INTRON A car cette combinaison fournit des taux de réponse sensiblement meilleurs [voir Etudes cliniques ].
- Les patients présentant les caractéristiques suivantes sont moins susceptibles de bénéficier d'un retraitement après l'échec d'un traitement : non-réponse antérieure, traitement antérieur par l'interféron pégylé, fibrose en pont importante ou cirrhose et infection de génotype 1 [voir Etudes cliniques ].
- Aucune donnée de sécurité et d'efficacité n'est disponible pour un traitement de plus d'un an.
DOSAGE ET ADMINISTRATION
Les gélules de REBETOL ne doivent en aucun cas être ouvertes, écrasées ou brisées. REBETOL 200 mg doit être pris avec de la nourriture [voir PHARMACOLOGIE CLINIQUE ]. REBETOL ne doit pas être utilisé chez les patients dont la clairance de la créatinine est inférieure à 50 mL/min.
Thérapie combinée REBETOL/PegIntron
Patients adultes
La dose recommandée de PegIntron est de 1,5 mcg/kg/semaine par voie sous-cutanée en association avec 800 à 1400 mg de REBETOL 200 mg par voie orale en fonction du poids corporel du patient (voir tableau 1). Le volume de PegIntron à injecter dépend de la force de PegIntron et du poids corporel du patient, reportez-vous à l'étiquetage de PegIntron pour des informations de dosage supplémentaires.
Durée du traitement – Patients naïfs à l'interféron alpha
La durée du traitement pour les patients de génotype 1 est de 48 semaines. L'arrêt du traitement doit être envisagé chez les patients qui n'obtiennent pas une chute ou une perte d'ARN-VHC d'au moins 2 log10 à 12 semaines, ou si l'ARN-VHC reste détectable après 24 semaines de traitement. Les patients de génotype 2 et 3 doivent être traités pendant 24 semaines.
Durée du traitement - Re-traitement avec PegIntron/REBETOL des échecs de traitement antérieurs
La durée du traitement pour les patients en échec thérapeutique antérieur est de 48 semaines, quel que soit le génotype du VHC. Les patients retraités qui ne parviennent pas à obtenir un ARN-VHC indétectable à la 12e semaine de traitement, ou dont l'ARN-VHC reste détectable après 24 semaines de traitement, sont très peu susceptibles d'obtenir une RVS et l'arrêt du traitement doit être envisagé [voir Etudes cliniques ].
Patients pédiatriques
Le dosage pour les patients pédiatriques est déterminé par la surface corporelle pour PegIntron et par le poids corporel pour REBETOL. La dose recommandée de PegIntron est de 60 mcg/m²/semaine sous-cutanée en association avec 15 mg/kg/jour de REBETOL 200 mg par voie orale en deux doses fractionnées (voir tableau 2) pour les patients pédiatriques âgés de 3 à 17 ans. Les patients qui atteignent leur 18e anniversaire alors qu'ils reçoivent PegIntron/REBETOL 200 mg doivent continuer à suivre le schéma posologique pédiatrique. La durée du traitement pour les patients de génotype 1 est de 48 semaines. Les patients de génotype 2 et 3 doivent être traités pendant 24 semaines.
REBETOL/INTRON A Thérapie combinée
Adultes
Durée du traitement – Patients naïfs à l'interféron alpha
La dose recommandée d'INTRON A est de 3 millions d'UI trois fois par semaine par voie sous-cutanée. La dose recommandée de capsules REBETOL dépend du poids corporel du patient (voir le tableau 3). La durée de traitement recommandée pour les patients non préalablement traités par interféron est de 24 à 48 semaines. La durée du traitement doit être adaptée au patient en fonction des caractéristiques initiales de la maladie, de la réponse au traitement et de la tolérance du traitement [voir INDICATIONS ET USAGE , EFFETS INDÉSIRABLES , et Etudes cliniques ]. Après 24 semaines de traitement, la réponse virologique doit être évaluée. L'arrêt du traitement doit être envisagé chez tout patient n'ayant pas atteint un ARN-VHC inférieur à la limite de détection du test à 24 semaines. Il n'y a pas de données de sécurité et d'efficacité sur un traitement de plus de 48 semaines dans la population de patients non traités précédemment.
Durée du traitement – Re-traitement avec INTRON A/REBETOL chez les patients en rechute
Chez les patients qui rechutent après une monothérapie par interféron non pégylé, la durée de traitement recommandée est de 24 semaines.
Pédiatrie
La dose recommandée de REBETOL est de 15 mg/kg par jour par voie orale (dose fractionnée AM et PM). Se référer à la Table 2 pour le Dosage de Pédiatrie de REBETOL dans la combinaison avec INTRON A. INTRON A pour l'Injection par le poids de corps de 25 kg à 61 kg est de 3 millions UI/m² trois fois par semaine sous-cutanément. Reportez-vous au tableau de dosage pour adultes pour un poids corporel supérieur à 61 kg.
La durée de traitement recommandée est de 48 semaines pour les patients pédiatriques de génotype 1. Après 24 semaines de traitement, la réponse virologique doit être évaluée. L'arrêt du traitement doit être envisagé chez tout patient qui n'a pas atteint un ARN-VHC inférieur à la limite de détection du test à ce moment. La durée de traitement recommandée pour les patients pédiatriques de génotype 2/3 est de 24 semaines.
Tests de laboratoire
Les tests de laboratoire suivants sont recommandés pour tous les patients traités avec REBETOL 200 mg, avant le début du traitement, puis périodiquement par la suite.
Tests hématologiques standard - y compris l'hémoglobine (prétraitement, semaine 2 et semaine 4 du traitement, et selon les besoins cliniques [voir AVERTISSEMENTS ET PRECAUTIONS ], numération complète et différentielle des globules blancs et numération plaquettaire.
- Chimie du sang - tests de la fonction hépatique et TSH.
- Grossesse - y compris le suivi mensuel pour les femmes en âge de procréer.
- ECG [voir AVERTISSEMENTS ET PRECAUTIONS ].
Modifications posologiques
Si des effets indésirables graves ou des anomalies de laboratoire se développent pendant le traitement par l'association REBETOL/INTRON A ou le traitement par REBETOL/PegIntron, modifiez ou arrêtez la dose jusqu'à ce que l'effet indésirable s'atténue ou diminue en gravité [voir AVERTISSEMENTS ET PRECAUTIONS ]. Si l'intolérance persiste après l'adaptation de la dose, le traitement combiné doit être arrêté. La réduction de dose de PegIntron dans les patients adultes sur la thérapie de combinaison REBETOL/PegIntron est accomplie dans un processus en deux étapes de la dose de départ originale de 1.5 mcg/kg/week, à 1 mcg/kg/week, puis à 0.5 mcg/kg/week , si besoin. Reportez-vous à l'étiquetage de PegIntron pour des informations supplémentaires concernant la réduction de la dose de PegIntron.
Dans l'Étude de thérapie de combinaison adulte 2, les réductions de dose se sont produites dans 42 % de sujets recevant PegIntron 1.5 mcg/kg et REBETOL 800 mgs tous les jours, en incluant 57 % de ces sujets pesant 60 kg ou moins. Dans l'étude 4, 16 % des sujets ont eu une réduction de dose de PegIntron à 1 mcg/kg en association avec REBETOL, avec 4 % supplémentaires nécessitant la deuxième réduction de dose de PegIntron à 0,5 mcg/kg en raison d'effets indésirables [voir EFFETS INDÉSIRABLES ].
La réduction de dose chez les patients pédiatriques est réalisée en modifiant la dose recommandée de PegIntron en deux étapes, de la dose initiale initiale de 60 mcg/m²/semaine, à 40 mcg/m²/semaine, puis à 20 mcg/m²/semaine, si nécessaire (voir tableau 4). Dans l'essai pédiatrique sur la polythérapie, des réductions de dose sont survenues chez 25 % des sujets recevant PegIntron 60 mcg/m² par semaine et REBETOL 15 mg/kg par jour. La réduction de dose dans les patients de pédiatrie est accomplie en modifiant la dose REBETOL recommandée de la dose de départ originale de 15 mgs/kg tous les jours dans un processus en deux étapes à 12 mgs/kg/jours, alors à 8 mgs/kg/jours, si nécessaire ( voir tableau 4).
REBETOL 200 mg ne doit pas être utilisé chez les patients dont la clairance de la créatinine est inférieure à 50 mL/min. Les patients présentant une fonction rénale altérée et ceux de plus de 50 ans doivent être étroitement surveillés en ce qui concerne le développement d'une anémie [voir AVERTISSEMENTS ET PRECAUTIONS , Utilisation dans des populations spécifiques , et PHARMACOLOGIE CLINIQUE ].
REBETOL 200 mg doit être administré avec prudence aux patients présentant une maladie cardiaque préexistante. Les patients doivent être évalués avant le début du traitement et doivent être surveillés de manière appropriée pendant le traitement. En cas de détérioration de l'état cardiovasculaire, le traitement doit être arrêté [voir AVERTISSEMENTS ET PRECAUTIONS ].
Pour les patients ayant des antécédents de maladie cardiovasculaire stable, une réduction permanente de la dose est nécessaire si l'hémoglobine diminue de plus ou égale à 2 g/dL au cours d'une période de 4 semaines. De plus, pour ces patients ayant des antécédents cardiaques, si l'hémoglobine reste inférieure à 12 g/dL après 4 semaines à dose réduite, le patient doit interrompre le traitement combiné.
Il est recommandé qu'un patient dont le taux d'hémoglobine tombe en dessous de 10 g/dL fasse modifier ou arrêter sa dose de REBETOL conformément au tableau 4 [voir AVERTISSEMENTS ET PRECAUTIONS ].
Reportez-vous à l'étiquetage d'INTRON A ou de PegIntron pour plus d'informations sur la façon de réduire une dose d'INTRON A ou de PegIntron.
Arrêt du dosage
Adultes
Chez les patients infectés par le VHC de génotype 1 et n'ayant jamais reçu d'interféron alfa recevant PegIntron en association avec la ribavirine, l'arrêt du traitement est recommandé s'il n'y a pas au moins une chute de 2 log10 ou une perte d'ARN-VHC à 12 semaines de traitement, ou si l'ARN-VHC les niveaux restent détectables après 24 semaines de traitement. Quel que soit le génotype, les patients précédemment traités qui ont un ARN-VHC détectable à la semaine 12 ou 24 sont très peu susceptibles d'obtenir une RVS et l'arrêt du traitement doit être envisagé.
Pédiatrie (3-17 ans)
Il est recommandé que les patients recevant l'association PegIntron/REBETOL (à l'exclusion du VHC de génotypes 2 et 3) soient interrompus du traitement à 12 semaines si leur ARN-VHC à la semaine 12 a chuté de moins de 2 log10 par rapport à un prétraitement ou à 24 semaines s'ils ont des symptômes détectables. ARN-VHC à la semaine de traitement 24.
COMMENT FOURNIE
Formes posologiques et points forts
REBETOL 200mg Gélules 200 mg
REBETOL 200mg Solution buvable 40 mg par mL
Stockage et manutention
REBETOL 200 mg Gélules sont des capsules blanches et opaques avec REBETOL, 200 mg, et le logo Schering Corporation imprimé sur l'enveloppe de la capsule ; les gélules sont conditionnées dans un flacon contenant 56 gélules ( CDN 0085-1351-05), 70 gélules ( CDN 0085-1385-07), et 84 gélules ( CDN 0085-1194-03).
REBETOL 200mg Solution buvable 40 mg par mL est un liquide clair, incolore à pâle ou jaune pâle à saveur de gomme à mâcher et il est emballé dans des bouteilles en verre ambré de 4 oz (100 mL/bouteille) avec des fermetures à l'épreuve des enfants ( CDN 0085-1318-01).
La bouteille de Capsules REBETOL devrait être conservée à 25°C (77°F); excursions autorisées jusqu'à 15-30°C (59-86°F) [voir Température ambiante contrôlée USP ].
La solution orale REBETOL 200 mg doit être conservée entre 2 et 8 °C (36 et 46 °F) ou à 25 °C (77 °F) ; excursions autorisées jusqu'à 15-30°C (59-86°F) [voir Température ambiante contrôlée USP ].
REBETOL 200mg Solution Orale fabriquée pour : Merck Sharp & Dohme Corp., une filiale de Whitehouse Station, NJ 08889, USA. Fabriqué par : Schering-Plough Canada, Inc., Pointe Claire, Québec, Canada Capsules REBETOL fabriquées par : Merck Sharp & Dohme Corp., une filiale de Whitehouse Station, NJ 08889, États-Unis. Révisé : décembre 2014.
EFFETS SECONDAIRES
Des essais cliniques avec REBETOL en association avec PegIntron ou INTRON A ont été menés chez plus de 7800 sujets âgés de 3 à 76 ans.
La principale toxicité de la ribavirine est l'anémie hémolytique. Des réductions des taux d'hémoglobine sont survenues au cours des 1 à 2 premières semaines de traitement par voie orale. Des réactions cardiaques et pulmonaires associées à l'anémie sont survenues chez environ 10 % des patients [voir AVERTISSEMENTS ET PRECAUTIONS ].
Plus de 96 % de tous les sujets des essais cliniques ont présenté un ou plusieurs effets indésirables. Les effets indésirables les plus fréquemment rapportés chez les sujets adultes recevant PegIntron ou INTRON A en association avec REBETOL 200 mg étaient une inflammation/réaction au site d'injection, fatigue/asthénie, maux de tête, frissons, fièvres, nausées, myalgie et anxiété/labilité émotionnelle/irritabilité. Les effets indésirables les plus fréquents chez les sujets pédiatriques, âgés de 3 ans et plus, recevant REBETOL 200 mg en association avec PegIntron ou INTRON A étaient la pyrexie, les maux de tête, la neutropénie, la fatigue, l'anorexie, l'érythème au site d'injection et les vomissements.
La section Effets indésirables fait référence aux essais cliniques suivants :
- Essais de thérapie combinée REBETOL/PegIntron :
- Étude clinique 1 - évaluation de la monothérapie par PegIntron (non décrite plus en détail dans cette étiquette ; voir l'étiquette de PegIntron pour plus d'informations sur cet essai).
- Étude 2 – a évalué REBETOL 800 mg/jour à plat en association avec 1,5 mcg/kg/semaine PegIntron ou avec INTRON A.
- Étude 3 – évaluation de PegIntron/REBETOL 200 mg basé sur le poids en association avec PegIntron/régime REBETOL à dose fixe de 200 mg.
- Étude 4 - a comparé deux doses de PegIntron (1,5 mcg/kg/semaine et 1 mcg/kg/semaine) en association avec REBETOL et un troisième groupe de traitement recevant Pegasys® (180 mcg/semaine)/Copegus® (1000-1200 mg/jour ).
- Étude 5 - PegIntron évalué (1,5 mcg/kg/semaine) en association avec REBETOL basé sur le poids chez des sujets en échec de traitement antérieur.
- Traitement combiné PegIntron/REBETOL 200 mg chez les patients pédiatriques
- Essais de thérapie combinée REBETOL/INTRON A pour adultes et pédiatrie
Des effets indésirables graves sont survenus chez environ 12 % des sujets dans les essais cliniques avec PegIntron avec ou sans REBETOL [voir AVERTISSEMENT ENCADRÉ , AVERTISSEMENTS ET PRECAUTIONS ]. Les événements graves les plus fréquents survenus chez les sujets traités par PegIntron et REBETOL 200 mg étaient la dépression et les idées suicidaires [voir AVERTISSEMENTS ET PRECAUTIONS ], chacun se produisant à une fréquence inférieure à 1 %. Les idées ou tentatives de suicide sont survenues plus fréquemment chez les patients pédiatriques, principalement les adolescents, par rapport aux patients adultes (2,4 % contre 1 %) pendant le traitement et le suivi hors traitement [voir AVERTISSEMENTS ET PRECAUTIONS ]. La réaction mortelle la plus fréquente survenant chez les sujets traités avec PegIntron et REBETOL 200 mg était un arrêt cardiaque, des idées suicidaires et une tentative de suicide [voir AVERTISSEMENTS ET PRECAUTIONS ], tous survenant chez moins de 1 % des sujets.
Étant donné que les essais cliniques sont menés dans des conditions très variables, les taux d'effets indésirables observés dans les essais cliniques d'un médicament ne peuvent pas être directement comparés aux taux des essais cliniques d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique clinique.
Expérience des essais cliniques - Thérapie combinée REBETOL/PegIntron
Sujets adultes
Les effets indésirables survenus au cours de l'essai clinique à une incidence supérieure à 5 % sont fournis par groupe de traitement de la thérapie combinée REBETOL/PegIntron (étude 2) dans le tableau 5.
Le tableau 6 résume les effets indésirables liés au traitement dans l'étude 4 qui se sont produits à une incidence supérieure ou égale à 10 %.
L'incidence des effets indésirables graves était comparable dans tous les essais. Dans l'Étude 3, il y avait une incidence semblable de réactions défavorables sérieuses annoncées pour le groupe REBETOL basé sur le poids (12 %) et pour le régime REBETOL de la dose plate. Dans l'Étude 2, l'incidence de réactions défavorables sérieuses était 17 % dans les groupes PegIntron/REBETOL comparés à 14 % dans le groupe INTRON A/REBETOL.
Dans de nombreux cas, mais pas tous, les effets indésirables ont disparu après réduction de la dose ou arrêt du traitement. Certains sujets ont présenté des effets indésirables graves en cours ou nouveaux au cours de la période de suivi de 6 mois. Dans l'étude 2, de nombreux sujets ont continué à présenter des effets indésirables plusieurs mois après l'arrêt du traitement. À la fin de la période de suivi de 6 mois, l'incidence des effets indésirables en cours par catégorie corporelle dans le groupe PegIntron 1,5/REBETOL 200 mg était de 33 % (psychiatrique), 20 % (musculo-squelettique) et 10 % (pour les troubles endocriniens et pour GI). Chez environ 10 à 15 % des sujets, la perte de poids, la fatigue et les maux de tête n'avaient pas disparu.
Il y a eu 31 décès de sujets survenus pendant le traitement ou pendant le suivi de ces essais cliniques. Dans l'étude 1, il y a eu 1 suicide chez un sujet recevant PegIntron en monothérapie et 2 décès parmi les sujets recevant INTRON A en monothérapie (1 meurtre/suicide et 1 mort subite). Dans l'étude 2, il y a eu 1 suicide chez un sujet recevant la thérapie combinée PegIntron/REBETOL 200 mg ; et 1 décès de sujet dans le groupe INTRON A/REBETOL 200 mg (accident de véhicule à moteur). Dans l'étude 3, il y a eu 14 décès, dont 2 étaient des suicides probables et 1 était un décès inexpliqué chez une personne ayant des antécédents médicaux pertinents de dépression. Dans l'étude 4, il y a eu 12 décès, dont 6 sont survenus chez des sujets ayant reçu la thérapie combinée PegIntron/REBETOL, 5 dans le bras PegIntron 1,5 mcg/REBETOL (N=1019) et 1 dans le bras PegIntron 1 mcg/REBETOL (N= 1016), et dont 6 sont survenus chez des sujets recevant Pegasys/Copegus (N = 1035) ; il y a eu 3 suicides qui se sont produits pendant la période de suivi sans traitement chez les sujets qui ont reçu la thérapie combinée PegIntron (1,5 mcg/kg)/REBETOL 200 mg.
Dans les études 1 et 2, 10 à 14 % des sujets recevant PegIntron, seul ou en association avec REBETOL 200 mg, ont arrêté le traitement contre 6 % traités avec INTRON A seul et 13 % traités avec INTRON A en association avec REBETOL. De même, dans l'étude 3, 15 % des sujets recevant PegIntron en association avec REBETOL basé sur le poids et 14 % des sujets recevant PegIntron et une dose plate de REBETOL 200 mg ont arrêté le traitement en raison d'un effet indésirable. Les raisons les plus courantes de l'arrêt du traitement étaient liées à des effets indésirables connus de l'interféron d'ordre psychiatrique, systémique (p. ex., fatigue, céphalées) ou gastro-intestinal. Dans l'étude 4, 13 % des sujets du bras PegIntron 1,5 mcg/REBETOL 200 mg, 10 % du bras PegIntron 1 mcg/REBETOL 200 mg et 13 % du bras Pegasys 180 mcg/Copegus ont arrêté en raison d'événements indésirables.
Dans l'Étude 2, les réductions de dose dues aux réactions défavorables se sont produites dans 42 % de sujets recevant PegIntron (1.5 mcg/kg)/REBETOL 200 mgs et dans 34 % de ceux recevant INTRON A/REBETOL. La majorité de sujets (57 %) pesant 60 kg ou moins recevant PegIntron (1.5 mcg/kg)/REBETOL ont exigé la réduction de dose. La réduction de l'interféron était liée à la dose (PegIntron 1,5 mcg/kg supérieur à PegIntron 0,5 mcg/kg ou INTRON A), 40 %, 27 %, 28 %, respectivement. La réduction de dose pour REBETOL 200 mg était similaire dans les trois groupes, 33 à 35 %. Les raisons les plus fréquentes des modifications de dose étaient la neutropénie (18 %) ou l'anémie (9 %) (voir Valeurs de laboratoire ). D'autres raisons courantes comprenaient la dépression, la fatigue, les nausées et la thrombocytopénie. Dans l'étude 3, les modifications de dose dues à des effets indésirables sont survenues plus fréquemment avec le dosage basé sur le poids (WBD) par rapport au dosage plat (29 % et 23 %, respectivement). Dans l'étude 4, 16 % des sujets ont eu une réduction de dose de PegIntron à 1 mcg/kg en association avec REBETOL 200 mg, avec 4 % supplémentaires nécessitant la deuxième réduction de dose de PegIntron à 0,5 mcg/kg en raison d'événements indésirables contre 15 % des sujets du bras Pegasys/Copegus, qui ont nécessité une réduction de dose à 135 mcg/semaine avec Pegasys, avec 7 % supplémentaires dans le bras Pegasys/Copegus nécessitant une deuxième réduction de dose à 90 mcg/semaine avec Pegasys.
Dans les essais sur l'association PegIntron/REBETOL 200 mg, les effets indésirables les plus courants étaient d'ordre psychiatrique, survenus chez 77 % des sujets de l'étude 2 et 68 % à 69 % des sujets de l'étude 3. Ces effets indésirables psychiatriques comprenaient le plus souvent la dépression, l'irritabilité et l'insomnie, chacune signalée par environ 30 % à 40 % des sujets dans tous les groupes de traitement. Des comportements suicidaires (idées, tentatives et suicides) sont survenus chez 2 % de tous les sujets pendant le traitement ou pendant le suivi après l'arrêt du traitement [voir AVERTISSEMENTS ET PRECAUTIONS ]. Dans l'étude 4, des effets indésirables psychiatriques sont survenus chez 58 % des sujets du bras PegIntron 1,5 mcg/REBETOL 200 mg, 55 % des sujets du bras PegIntron 1 mcg/REBETOL 200 mg et 57 % des sujets du bras Pegasys 180 mcg/Copegus .
PegIntron a induit de la fatigue ou des maux de tête chez environ les deux tiers des sujets, avec de la fièvre ou des frissons chez environ la moitié des sujets. La sévérité de certains de ces symptômes systémiques (p. ex., fièvre et céphalées) avait tendance à diminuer à mesure que le traitement se poursuivait. Dans les études 1 et 2, l'inflammation et la réaction au site d'application (par exemple, ecchymoses, démangeaisons et irritation) sont survenues à environ deux fois l'incidence avec les thérapies PegIntron (chez jusqu'à 75 % des sujets) par rapport à INTRON A. Cependant, la douleur au site d'injection était peu fréquent (2 à 3 %) dans tous les groupes. Dans l'étude 3, il y avait une incidence globale de 23 % à 24 % pour les réactions au site d'injection ou l'inflammation.
Les sujets recevant REBETOL/PegIntron en retraitement après l'échec d'un précédent régime d'association d'interféron ont signalé des effets indésirables similaires à ceux précédemment associés à ce régime au cours des essais cliniques sur des sujets naïfs de traitement.
Sujets pédiatriques
En général, le profil des effets indésirables dans la population pédiatrique était similaire à celui observé chez les adultes. Dans l'essai pédiatrique, les effets indésirables les plus fréquents chez tous les sujets étaient la pyrexie (80 %), les céphalées (62 %), la neutropénie (33 %), la fatigue (30 %), l'anorexie (29 %), l'érythème au point d'injection (29 %). %) et des vomissements (27 %). La majorité des effets indésirables signalés au cours de l'essai étaient d'intensité légère ou modérée. Des effets indésirables graves ont été signalés chez 7 % (8/107) de tous les sujets et comprenaient des douleurs au point d'injection (1 %), des douleurs aux extrémités (1 %), des maux de tête (1 %), de la neutropénie (1 %) et de la pyrexie (4 %). Les effets indésirables importants survenus dans cette population de sujets étaient la nervosité (7 % ; 7/107), l'agressivité (3 % ; 3/107), la colère (2 % ; 2/107) et la dépression (1 % ; 1/107) . Cinq sujets ont reçu un traitement à la lévothyroxine, trois avec une hypothyroïdie clinique et deux avec des élévations asymptomatiques de la TSH. Le gain de poids et de taille des sujets pédiatriques traités avec PegIntron plus REBETOL était en retard par rapport à celui prédit par les données de population normatives pour toute la durée du traitement. Une vitesse de croissance fortement inhibée (inférieure au 3e centile) a été observée chez 70 % des sujets pendant le traitement.
Des modifications de dose de PegIntron et/ou de ribavirine ont été nécessaires chez 25 % des sujets en raison d'effets indésirables liés au traitement, le plus souvent pour l'anémie, la neutropénie et la perte de poids. Deux sujets (2 % ; 2/107) ont arrêté le traitement en raison d'un effet indésirable.
Les effets indésirables survenus avec une incidence supérieure ou égale à 10 % chez les sujets pédiatriques de l'essai sont présentés dans le tableau 7.
Quatre-vingt-quatorze des 107 sujets inscrits à un essai de suivi à long terme de 5 ans. Les effets à long terme sur la croissance étaient moindres chez les sujets traités pendant 24 semaines que chez ceux traités pendant 48 semaines. Vingt-quatre pour cent des sujets (11/46) traités pendant 24 semaines et 40 % des sujets (19/48) traités pendant 48 semaines ont présenté une diminution de la taille pour l'âge > 15 percentiles entre le prétraitement et la fin de la période de 5 ans. suivi à long terme par rapport aux centiles de référence avant le traitement. Onze pour cent des sujets (5/46) traités pendant 24 semaines et 13 % des sujets (6/48) traités pendant 48 semaines ont présenté une diminution par rapport à la ligne de base avant le traitement de > 30 percentiles de taille pour l'âge jusqu'à la fin du suivi à long terme de 5 ans. Bien qu'observé dans tous les groupes d'âge, le risque le plus élevé de diminution de la taille à la fin du suivi à long terme semblait être corrélé à l'initiation d'un traitement combiné au cours des années de vitesse de croissance maximale attendue. [Voir AVERTISSEMENTS ET PRECAUTIONS ]
Valeurs de laboratoire
Sujets adultes et pédiatriques
Le profil d'effets indésirables de l'étude 3, qui comparait l'association PegIntron/REBETOL 200 mg en fonction du poids à un régime PegIntron/REBETOL à dose fixe de 200 mg, a révélé un taux accru d'anémie avec une posologie en fonction du poids (29 % contre 19 % pour le dosage en fonction du poids). vs régimes à dose fixe, respectivement). Cependant, la majorité des cas d'anémie étaient bénins et répondaient aux réductions de dose.
Les modifications des valeurs de laboratoire sélectionnées pendant le traitement en association avec le traitement REBETOL 200 mg sont décrites ci-dessous. Les diminutions de l'hémoglobine, des leucocytes, des neutrophiles et des plaquettes peuvent nécessiter une réduction de la dose ou l'arrêt définitif du traitement [voir DOSAGE ET ADMINISTRATION ]. Les changements dans les valeurs de laboratoire sélectionnées pendant le traitement sont décrits dans le tableau 8. La plupart des changements dans les valeurs de laboratoire dans l'essai PegIntron/REBETOL 200 mg avec la pédiatrie étaient légers ou modérés.
Hémoglobine
Les taux d'hémoglobine ont diminué à moins de 11 g/dL chez environ 30 % des sujets de l'étude 2. Dans l'étude 3, 47 % des sujets recevant WBD REBETOL 200 mg et 33 % sous dose plate REBETOL 200 mg ont présenté une diminution du taux d'hémoglobine inférieure à 11 g. /dl. Les réductions de l'hémoglobine à moins de 9 g/dL sont survenues plus fréquemment chez les sujets recevant une WBD par rapport à une dose fixe (4 % et 2 %, respectivement). Dans l'étude 2, une modification de la dose a été nécessaire chez 9 % et 13 % des sujets des groupes PegIntron/REBETOL 200 mg et INTRON A/REBETOL 200 mg. Dans l'étude 4, les sujets recevant PegIntron (1,5 mcg/kg)/REBETOL ont présenté des diminutions du taux d'hémoglobine entre 8,5 et moins de 10 g/dL (28 %) et moins de 8,5 g/dL (3 %), alors que chez les patients recevant Pegasys 180 mcg/Copegus, ces diminutions sont survenues chez 26 % et 4 % des sujets, respectivement. Les taux d'hémoglobine sont devenus stables après le traitement des semaines 4 à 6 en moyenne. Le schéma typique observé était une diminution des taux d'hémoglobine par la semaine de traitement 4 suivie d'une stabilisation et d'un plateau, qui s'est maintenu jusqu'à la fin du traitement. Dans l'essai de monothérapie PegIntron, les diminutions d'hémoglobine étaient généralement légères et des modifications de dose étaient rarement nécessaires [voir DOSAGE ET ADMINISTRATION ].
Neutrophiles
Des diminutions du nombre de neutrophiles ont été observées chez la majorité des sujets adultes traités par une thérapie combinée avec REBETOL 200 mg dans l'étude 2 (85 %) et INTRON A/REBETOL (60 %). Une neutropénie sévère potentiellement mortelle (moins de 0,5 x 109/L) est survenue chez 2 % des sujets traités par INTRON A/REBETOL 200 mg et chez environ 4 % des sujets traités par PegIntron/REBETOL 200 mg dans l'étude 2. Dix-huit pour cent des sujets recevant PegIntron/REBETOL dans l'étude 2 a nécessité une modification de la posologie de l'interféron. Peu de sujets (moins de 1 %) ont nécessité un arrêt définitif du traitement. Le nombre de neutrophiles est généralement revenu aux niveaux d'avant le traitement 4 semaines après l'arrêt du traitement [voir DOSAGE ET ADMINISTRATION ].
Plaquettes
Le nombre de plaquettes a diminué à moins de 100 000/mm³ chez environ 20 % des sujets traités avec PegIntron seul ou avec REBETOL 200 mg et chez 6 % des sujets adultes traités avec INTRON A/REBETOL. Des diminutions sévères du nombre de plaquettes (moins de 50 000/mm³) surviennent chez moins de 4 % des sujets adultes. Les patients peuvent nécessiter un arrêt ou une modification de la dose en raison de la diminution des plaquettes [voir DOSAGE ET ADMINISTRATION ]. Dans l'étude 2, 1 % ou 3 % des sujets ont nécessité une modification de la dose d'INTRON A ou de PegIntron, respectivement. La numération plaquettaire est généralement revenue aux niveaux de prétraitement 4 semaines après l'arrêt du traitement.
La fonction thyroïdienne
Le développement d'anomalies de la TSH, avec ou sans manifestations cliniques, est associé aux traitements par interféron. Dans l'Étude 2, les désordres thyroïdiens cliniquement apparents se sont produits parmi les sujets traités avec INTRON A ou PegIntron (avec ou sans REBETOL) à une incidence semblable (5 % pour hypothyroidism et 3 % pour hyperthyroïdism). Les sujets ont développé de nouvelles anomalies de la TSH pendant le traitement et pendant la période de suivi. À la fin de la période de suivi, 7 % des sujets avaient encore des valeurs de TSH anormales.
Bilirubine et acide urique
Dans l'étude 2, 10 à 14 % des sujets ont développé une hyperbilirubinémie et 33 à 38 % ont développé une hyperuricémie en association avec une hémolyse. Six sujets ont développé une goutte légère à modérée.
Expérience des essais cliniques – Thérapie combinée REBETOL/INTRON A
Sujets adultes
Dans les essais cliniques, 19 % et 6 % des sujets précédemment non traités et des sujets en rechute, respectivement, ont interrompu le traitement en raison d'effets indésirables dans les bras combinés, contre 13 % et 3 % dans les bras interféron. Les effets indésirables sélectionnés liés au traitement qui se sont produits dans les essais américains avec une incidence supérieure ou égale à 5 % sont fournis par groupe de traitement (voir le tableau 9). En général, les effets indésirables sélectionnés liés au traitement ont été signalés avec une incidence plus faible dans les essais internationaux par rapport aux essais américains, à l'exception de l'asthénie, des symptômes pseudo-grippaux, de la nervosité et du prurit.
Sujets pédiatriques
Lors d'essais cliniques sur 118 sujets pédiatriques âgés de 3 à 16 ans, 6 % ont arrêté le traitement en raison d'effets indésirables. Des modifications de dose ont été nécessaires chez 30 % des sujets, le plus souvent pour anémie et neutropénie. En général, le profil des effets indésirables dans la population pédiatrique était similaire à celui observé chez les adultes. Les troubles au site d'injection, la fièvre, l'anorexie, les vomissements et la labilité émotionnelle sont survenus plus fréquemment chez les sujets pédiatriques que chez les sujets adultes. À l'inverse, les sujets pédiatriques ont présenté moins de fatigue, de dyspepsie, d'arthralgie, d'insomnie, d'irritabilité, de troubles de la concentration, de dyspnée et de prurit que les sujets adultes. Les effets indésirables sélectionnés liés au traitement qui se sont produits avec une incidence supérieure ou égale à 5 % parmi tous les sujets pédiatriques qui ont reçu la dose recommandée de thérapie combinée REBETOL/INTRON A sont fournis dans le tableau 9.
Au cours d'un traitement de 48 semaines, il y a eu une diminution du taux de croissance linéaire (diminution moyenne de l'attribution du centile de 7 %) et une diminution du taux de gain de poids (diminution moyenne de l'attribution du centile de 9 %). Une inversion générale de ces tendances a été notée au cours de la période post-traitement de 24 semaines. Les données à long terme chez un nombre limité de patients suggèrent cependant que la thérapie combinée peut induire une inhibition de la croissance qui se traduit par une taille adulte finale réduite chez certains patients [voir AVERTISSEMENTS ET PRECAUTIONS ].
Valeurs de laboratoire
Les modifications des valeurs hématologiques sélectionnées (hémoglobine, globules blancs, neutrophiles et plaquettes) au cours du traitement sont décrites ci-dessous (voir tableau 10).
Hémoglobine. Les diminutions d'hémoglobine parmi les sujets recevant la thérapie REBETOL ont commencé à la semaine 1, avec la stabilisation par la semaine 4. essai. Chez les sujets en rechute, la diminution maximale moyenne par rapport au départ était de 2,8 g/dL dans l'essai américain et de 2,6 g/dL dans l'essai international. Les valeurs d'hémoglobine sont revenues aux niveaux de prétraitement dans les 4 à 8 semaines suivant l'arrêt du traitement chez la plupart des sujets.
Bilirubine et acide urique. Des augmentations de la bilirubine et de l'acide urique, associées à l'hémolyse, ont été notées dans les essais cliniques. La plupart étaient des changements biochimiques modérés et ont été inversés dans les 4 semaines suivant l'arrêt du traitement. Cette observation s'est produite le plus fréquemment chez les sujets ayant déjà reçu un diagnostic de syndrome de Gilbert. Cela n'a pas été associé à un dysfonctionnement hépatique ou à une morbidité clinique.
Expériences post-commercialisation
Les effets indésirables suivants ont été identifiés et signalés lors de l'utilisation post-approbation de REBETOL en association avec INTRON A ou PegIntron. Étant donné que ces réactions sont signalées volontairement par une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale avec l'exposition au médicament.
Troubles du système sanguin et lymphatique
Aplasie pure des globules rouges, anémie aplasique
Troubles de l'oreille et du labyrinthe
Trouble auditif, vertige
Troubles respiratoires, thoraciques et médiastinaux
Hypertension pulmonaire
Troubles oculaires
Décollement séreux de la rétine
Troubles endocriniens
Diabète
INTERACTIONS MÉDICAMENTEUSES
Didanosine
L'exposition à la didanosine ou à son métabolite actif (didésoxyadénosine 5'-triphosphate) est augmentée lorsque la didanosine est co-administrée avec la ribavirine, ce qui pourrait provoquer ou aggraver des toxicités cliniques ; par conséquent, la co-administration de capsules ou de solution buvable REBETOL 200 mg et de didanosine est contre-indiquée. Des cas d'insuffisance hépatique mortelle, ainsi que de neuropathie périphérique, de pancréatite et d'hyperlactatémie/acidose lactique symptomatique ont été rapportés dans les essais cliniques.
Analogues nucléosidiques
Une décompensation hépatique (certaines mortelles) est survenue chez des patients cirrhotiques co-infectés par le VIH/VHC recevant une thérapie antirétrovirale combinée pour le VIH et l'interféron alpha et la ribavirine. L'ajout d'un traitement par interférons alpha seuls ou en association avec la ribavirine peut augmenter le risque dans cette population de patients. Les patients recevant de l'interféron avec de la ribavirine et des inhibiteurs nucléosidiques de la transcriptase inverse (INTI) doivent être étroitement surveillés pour les toxicités associées au traitement, en particulier la décompensation hépatique et l'anémie. L'arrêt des INTI doit être considéré comme médicalement approprié (voir étiquetage pour chaque produit INTI ). Une réduction de la dose ou l'arrêt de l'interféron, de la ribavirine ou des deux doivent également être envisagés si une aggravation des toxicités cliniques est observée, y compris une décompensation hépatique (p. ex., Child-Pugh supérieur à 6).
La ribavirine peut antagoniser l'activité antivirale en culture cellulaire de la stavudine et de la zidovudine contre le VIH. Il a été démontré que la ribavirine en culture cellulaire inhibe la phosphorylation de la lamivudine, de la stavudine et de la zidovudine, ce qui pourrait entraîner une diminution de l'activité antirétrovirale. Cependant, dans une étude avec un autre interféron pégylé en association avec la ribavirine, aucune interaction pharmacocinétique (p. ex., concentrations plasmatiques ou concentrations de métabolites actifs triphosphorylés intracellulaires) ou pharmacodynamique (p. ex., perte de la suppression virologique du VIH/VHC) n'a été observée lorsque la ribavirine et la lamivudine (n = 18), la stavudine (n = 10) ou la zidovudine (n = 6) ont été co-administrés dans le cadre d'un traitement multimédicamenteux chez des sujets co-infectés par le VIH/VHC. Par conséquent, l'utilisation concomitante de ribavirine avec l'un ou l'autre de ces médicaments doit être utilisée avec prudence.
Médicaments métabolisés par le cytochrome P-450
Les résultats d'études in vitro utilisant des préparations de microsomes de foie humain et de rat ont indiqué peu ou pas de métabolisme de la ribavirine médié par l'enzyme cytochrome P-450, avec un potentiel minimal d'interactions médicamenteuses basées sur l'enzyme P-450.
Aucune interaction pharmacocinétique n'a été notée entre INTRON A et les gélules de REBETOL 200 mg dans une étude pharmacocinétique à doses multiples.
Azathioprine
Il a été rapporté que l'utilisation de la ribavirine pour le traitement de l'hépatite C chronique chez les patients recevant de l'azathioprine induisait une pancytopénie sévère et pouvait augmenter le risque de myélotoxicité liée à l'azathioprine. L'inosine monophosphate déshydrogénase (IMDH) est nécessaire pour l'une des voies métaboliques de l'azathioprine. La ribavirine est connue pour inhiber l'IMDH, entraînant ainsi l'accumulation d'un métabolite de l'azathioprine, le 6-méthylthioinosine monophosphate (6-MTITP), qui est associé à une myélotoxicité (neutropénie, thrombocytopénie et anémie). Les patients recevant de l'azathioprine avec de la ribavirine doivent subir une numération globulaire complète, y compris une numération plaquettaire, surveillée chaque semaine pendant le premier mois, deux fois par mois pendant les deuxième et troisième mois de traitement, puis une fois par mois ou plus fréquemment si la posologie ou d'autres changements de traitement sont nécessaires [voir AVERTISSEMENTS ET PRECAUTIONS ].
AVERTISSEMENTS
Inclus dans le cadre du PRÉCAUTIONS section.
PRÉCAUTIONS
Grossesse
REBETOL 200 mg gélules et solution buvable peut entraîner des malformations congénitales et la mort de l'enfant à naître. La thérapie REBETOL ne devrait pas être commencée jusqu'à ce qu'un rapport d'un test de grossesse négatif ait été obtenu juste avant l'initiation planifiée de thérapie. Les patientes doivent utiliser au moins deux formes de contraception et subir des tests de grossesse mensuels pendant le traitement et pendant les 6 mois suivant l'arrêt du traitement. Des précautions extrêmes doivent être prises pour éviter une grossesse chez les patientes et chez les partenaires féminines des patients masculins. REBETOL 200mg a démontré effets tératogènes et embryocides significatifs chez toutes les espèces animales pour lesquelles des études adéquates ont été menées. Ces effets se sont produits à des doses aussi faible qu'un vingtième de la dose humaine recommandée de ribavirine. Le traitement par REBETOL 200 mg ne doit pas être commencé avant le rapport d'un test de grossesse négatif a été obtenu immédiatement avant le début prévu du traitement [voir AVERTISSEMENT ENCADRÉ , CONTRE-INDICATIONS , Utilisation dans des populations spécifiques , et INFORMATIONS PATIENTS ].
Anémie
La toxicité principale de la ribavirine est l'anémie hémolytique, qui a été observée chez environ 10 % des sujets traités par REBETOL/INTRON A dans les essais cliniques. L'anémie associée aux capsules REBETOL survient dans les 1 à 2 semaines suivant le début du traitement. Étant donné que la baisse initiale de l'hémoglobine peut être importante, il est conseillé d'obtenir l'hémoglobine ou l'hématocrite avant le début du traitement et à la semaine 2 et à la semaine 4 du traitement, ou plus fréquemment si cliniquement indiqué. Les patients doivent ensuite être suivis comme cliniquement approprié [voir DOSAGE ET ADMINISTRATION ].
Des infarctus du myocarde mortels et non mortels ont été signalés chez des patients souffrant d'anémie causée par REBETOL. Les patients doivent être évalués pour une maladie cardiaque sous-jacente avant le début du traitement par la ribavirine. Les patients présentant une maladie cardiaque préexistante doivent subir des électrocardiogrammes avant le traitement et doivent être surveillés de manière appropriée pendant le traitement. En cas de détérioration de l'état cardiovasculaire, le traitement doit être suspendu ou interrompu [voir DOSAGE ET ADMINISTRATION ]. Étant donné que la maladie cardiaque peut être aggravée par une anémie d'origine médicamenteuse, les patients ayant des antécédents de maladie cardiaque importante ou instable ne doivent pas utiliser REBETOL.
Pancréatite
Le traitement par REBETOL et INTRON A ou PegIntron doit être suspendu chez les patients présentant des signes et symptômes de pancréatite et interrompu chez les patients présentant une pancréatite confirmée.
Troubles pulmonaires
Les symptômes pulmonaires, en incluant la dyspnée, les infiltrats pulmonaires, la pneumonite, l'hypertension pulmonaire et la pneumonie, ont été annoncés pendant la thérapie avec REBETOL avec la thérapie de combinaison d'interféron alpha; des cas occasionnels de pneumonie mortelle se sont produits. De plus, la sarcoïdose ou l'exacerbation de la sarcoïdose a été rapportée. S'il existe des signes d'infiltrats pulmonaires ou d'altération de la fonction pulmonaire, le patient doit être étroitement surveillé et, le cas échéant, le traitement combiné doit être interrompu.
Troubles ophtalmologiques
La ribavirine est utilisée en association avec les interférons alpha. Diminution ou perte de la vision, rétinopathie, y compris œdème maculaire, artère ou veine rétinienne, thrombose, hémorragies rétiniennes et taches cotonneuses, névrite optique, œdème papillaire et décollement séreux de la rétine sont induits ou aggravés par un traitement aux interférons alpha. Tous les patients doivent subir un examen de la vue au départ. Les patients présentant des troubles ophtalmologiques préexistants (p. ex., rétinopathie diabétique ou hypertensive) doivent subir des examens ophtalmologiques périodiques pendant le traitement combiné avec un traitement par interféron alpha. Tout patient qui développe des symptômes oculaires doit subir un examen oculaire rapide et complet. Le traitement combiné avec les interférons alpha doit être interrompu chez les patients qui développent des troubles ophtalmologiques nouveaux ou qui s'aggravent.
Tests de laboratoire
PegIntron en association avec la ribavirine peut entraîner une diminution sévère du nombre de neutrophiles et de plaquettes, ainsi que des anomalies hématologiques, endocriniennes (par exemple, TSH) et hépatiques.
Les patients sous traitement combiné PegIntron/REBETOL doivent subir des tests d'hématologie et de biochimie sanguine avant le début du traitement, puis périodiquement par la suite. Dans l'essai clinique chez l'adulte, la numération globulaire complète (y compris la numération de l'hémoglobine, des neutrophiles et des plaquettes) et la chimie (y compris l'AST, l'ALT, la bilirubine et l'acide urique) ont été mesurées pendant la période de traitement aux semaines 2, 4, 8, 12 et puis à intervalles de 6 semaines, ou plus fréquemment si des anomalies se sont développées. Chez les sujets pédiatriques, les mêmes paramètres de laboratoire ont été évalués avec une évaluation supplémentaire de l'hémoglobine à la semaine de traitement 6. Les taux de TSH ont été mesurés toutes les 12 semaines pendant la période de traitement. L'ARN-VHC doit être mesuré périodiquement pendant le traitement [voir DOSAGE ET ADMINISTRATION ].
Troubles dentaires et parodontaux
Des troubles dentaires et parodontaux ont été rapportés chez des patients recevant un traitement combiné à la ribavirine et à l'interféron ou au peginterféron. De plus, la bouche sèche pourrait avoir un effet néfaste sur les dents et les muqueuses de la bouche lors d'un traitement à long terme avec l'association de REBETOL et d'interféron alfa-2b pégylé ou non pégylé. Les patients doivent se brosser soigneusement les dents deux fois par jour et subir des examens dentaires réguliers. En cas de vomissements, il faut leur conseiller de se rincer soigneusement la bouche par la suite.
Administration concomitante d'azathioprine
Une pancytopénie (diminution marquée des globules rouges, des neutrophiles et des plaquettes) et une suppression de la moelle osseuse ont été signalées dans la littérature comme se produisant dans les 3 à 7 semaines suivant l'administration concomitante d'interféron pégylé/ribavirine et d'azathioprine. Chez ce nombre limité de patients (n = 8), la myélotoxicité était réversible dans les 4 à 6 semaines après l'arrêt du traitement antiviral contre le VHC et de l'azathioprine concomitante et ne réapparaissait pas lors de la réintroduction de l'un ou l'autre traitement seul. PegIntron, REBETOL et l'azathioprine doivent être interrompus en cas de pancytopénie, et l'interféron pégylé/ribavirine ne doit pas être réintroduit avec l'azathioprine concomitante [voir INTERACTIONS MÉDICAMENTEUSES ].
Impact sur la croissance - Utilisation pédiatrique
Les données sur les effets de PegIntron et de REBETOL sur la croissance proviennent d'une étude ouverte chez des sujets âgés de 3 à 17 ans, dans laquelle les changements de poids et de taille sont comparés aux données normatives de la population américaine. En général, le gain de poids et de taille des sujets pédiatriques traités avec PegIntron et REBETOL 200 mg est inférieur à celui prédit par les données de population normatives pour toute la durée du traitement. Une vitesse de croissance fortement inhibée (inférieure au 3e centile) a été observée chez 70 % des sujets pendant le traitement. Après le traitement, un rebond de croissance et un gain de poids se sont produits chez la plupart des sujets. Les données de suivi à long terme chez les sujets pédiatriques indiquent cependant que PegIntron en association avec REBETOL peut induire une inhibition de la croissance qui entraîne une réduction de la taille adulte chez certains patients [voir EFFETS INDÉSIRABLES ].
De même, un impact sur la croissance a été observé chez les sujets après un traitement par REBETOL 200 mg et INTRON A en association pendant un an. Dans un essai de suivi à long terme d'un nombre limité de ces sujets, la thérapie combinée a entraîné une taille adulte finale réduite chez certains sujets [voir EFFETS INDÉSIRABLES ].
Garanties d'utilisation
D'après les résultats des essais cliniques, la ribavirine en monothérapie n'est pas efficace pour le traitement de l'infection chronique par le virus de l'hépatite C ; par conséquent, REBETOL 200 mg gélules ou solution buvable ne doit pas être utilisé seul. L'innocuité et l'efficacité des gélules et de la solution buvable REBETOL n'ont été établies que lorsqu'elles sont utilisées avec INTRON A ou PegIntron (pas d'autres interférons) en tant que thérapie combinée.
L'innocuité et l'efficacité de la thérapie REBETOL/INTRON A et PegIntron pour le traitement de l'infection par le VIH, l'adénovirus, le RSV, le parainfluenza ou les infections grippales n'ont pas été établies. REBETOL 200mg gélules ne doit pas être utilisé dans ces indications. La ribavirine pour inhalation a un étiquetage distinct, qui doit être consulté si un traitement par inhalation de ribavirine est envisagé.
Il existe des effets indésirables importants causés par la thérapie REBETOL/INTRON A ou PegIntron, y compris la dépression sévère et les idées suicidaires, l'anémie hémolytique, la suppression de la fonction de la moelle osseuse, les troubles auto-immuns et infectieux, le dysfonctionnement pulmonaire, la pancréatite et le diabète. Les idées ou tentatives de suicide sont survenues plus fréquemment chez les patients pédiatriques, principalement les adolescents, par rapport aux patients adultes (2,4 % contre 1 %) pendant le traitement et le suivi hors traitement. L'étiquetage d'INTRON A et de PegIntron doit être revu dans son intégralité pour des informations de sécurité supplémentaires avant le début du traitement combiné.
Informations sur les conseils aux patients
Conseillez au patient de lire l'étiquetage patient approuvé par la FDA ( Guide des médicaments ).
Anémie
L'effet indésirable le plus courant survenu avec les capsules REBETOL est l'anémie, qui peut être grave [voir AVERTISSEMENTS ET PRECAUTIONS et EFFETS INDÉSIRABLES ]. Les patients doivent être informés que des évaluations de laboratoire sont nécessaires avant de commencer le traitement et périodiquement par la suite [voir DOSAGE ET ADMINISTRATION ]. Il est conseillé aux patients de bien s'hydrater, en particulier pendant les premières étapes du traitement.
Grossesse
Les patients doivent être informés que REBETOL 200mg gélules et solution buvable peut provoquer des malformations congénitales et la mort de l'enfant à naître. REBETOL 200 mg ne doit pas être utilisé par les femmes enceintes ou par les hommes dont la partenaire féminine est enceinte. Des précautions extrêmes doivent être prises pour éviter une grossesse chez les patientes et chez les partenaires féminines de patients masculins prenant REBETOL. REBETOL ne doit pas être initié tant qu'un rapport de test de grossesse négatif n'a pas été obtenu immédiatement avant le début du traitement. Les patientes doivent effectuer un test de grossesse tous les mois pendant le traitement et pendant 6 mois après le traitement.
Les femmes en âge de procréer doivent être conseillées sur l'utilisation d'une contraception efficace (deux formes fiables) avant de commencer le traitement. Les patients (hommes et femmes) doivent être informés des risques tératogènes/embryocides et doivent être informés de la pratique d'une contraception efficace pendant REBETOL 200 mg et pendant 6 mois après le traitement. Les patients (hommes et femmes) doivent être informés qu'ils doivent informer immédiatement le médecin en cas de grossesse [voir CONTRE-INDICATIONS , AVERTISSEMENTS ET PRECAUTIONS , et Utilisation dans des populations spécifiques ].
Si une grossesse survient pendant le traitement ou pendant 6 mois après le traitement, la patiente doit être informée du risque tératogène du traitement par REBETOL 200 mg pour le fœtus. Les patientes ou les partenaires des patientes doivent immédiatement signaler à leur médecin toute grossesse survenue pendant le traitement ou dans les 6 mois suivant l'arrêt du traitement. Les prescripteurs doivent signaler ces cas en appelant le 1-800-593-2214.
Risques versus avantages
Les patients recevant des gélules REBETOL doivent être informés des avantages et des risques associés au traitement, dirigés dans son utilisation appropriée et référés au patient Guide des médicaments . Les patients doivent être informés que l'effet du traitement de l'hépatite C sur la transmission n'est pas connu et que des précautions appropriées pour prévenir la transmission du virus de l'hépatite C doivent être prises.
Les patients doivent être informés de ce qu'il faut faire s'ils oublient une dose de REBETOL ; la dose oubliée doit être prise dès que possible au cours de la même journée. Les patients ne doivent pas doubler la dose suivante. Les patients doivent être invités à contacter leur fournisseur de soins de santé s'ils ont des questions.
Toxicologie non clinique
Carcinogenèse, mutagenèse, altération de la fertilité
Carcinogenèse
La ribavirine n'a provoqué d'augmentation d'aucun type de tumeur lorsqu'elle a été administrée pendant 6 mois dans le modèle de souris transgénique déficiente en p53 à des doses allant jusqu'à 300 mg/kg (équivalent humain estimé de 25 mg/kg sur la base d'un ajustement de la surface corporelle pour un adulte de 60 kg). ; environ 1,9 fois la dose quotidienne maximale recommandée chez l'homme). La ribavirine s'est avérée non cancérogène lorsqu'elle a été administrée pendant 2 ans à des rats à des doses allant jusqu'à 40 mg/kg (équivalent humain estimé à 5,71 mg/kg sur la base d'un ajustement de la surface corporelle pour un adulte de 60 kg).
Mutagenèse
La ribavirine a démontré des incidences accrues de mutation et de transformation cellulaire dans de multiples tests de génotoxicité. La ribavirine était active dans le test de transformation cellulaire in vitro Balb/3T3. Une activité mutagène a été observée dans le test du lymphome de souris et à des doses de 20 à 200 mg/kg (équivalent humain estimé de 1,67 à 16,7 mg/kg, basé sur l'ajustement de la surface corporelle pour un adulte de 60 kg ; 0,1 à 1 fois la dose maximale dose humaine recommandée de ribavirine sur 24 heures) dans un test du micronoyau chez la souris. Un test de létalité dominante chez le rat était négatif, indiquant que si des mutations se produisaient chez le rat, elles n'étaient pas transmises par les gamètes mâles.
Altération de la fertilité
La ribavirine a démontré des effets embryocides et tératogènes significatifs à des doses bien inférieures à la dose humaine recommandée chez toutes les espèces animales chez lesquelles des études adéquates ont été menées. Des malformations du crâne, du palais, des yeux, de la mâchoire, des membres, du squelette et du tractus gastro-intestinal ont été notées. L'incidence et la gravité des effets tératogènes ont augmenté avec l'augmentation de la dose de médicament. La survie des fœtus et de la progéniture a été réduite. Dans les études conventionnelles d'embryotoxicité/tératogénicité chez le rat et le lapin, les niveaux de dose sans effet observés étaient bien inférieurs à ceux pour l'utilisation clinique proposée (0,3 mg/kg/jour pour le rat et le lapin ; environ 0,06 fois la dose recommandée chez l'homme sur 24 heures de ribavirine). Aucune toxicité maternelle ou effet sur la progéniture n'a été observé dans une étude de toxicité péri/postnatale chez des rats ayant reçu par voie orale jusqu'à 1 mg/kg/jour (dose équivalente humaine estimée de 0,17 mg/kg basée sur l'ajustement de la surface corporelle pour un adulte de 60 kg ; environ 0,01 fois la dose maximale recommandée de ribavirine sur 24 heures chez l'homme) [voir CONTRE-INDICATIONS , et AVERTISSEMENTS ET PRECAUTIONS ].
Les femmes fertiles et les partenaires de femmes fertiles ne doivent pas recevoir REBETOL sauf si la patiente et son partenaire utilisent une contraception efficace (deux formes fiables). Sur la base d'une demi-vie de doses multiples (t1/2) de ribavirine de 12 jours, une contraception efficace doit être utilisée pendant 6 mois après le traitement (par exemple, 15 demi-vies de clairance pour la ribavirine).
REBETOL doit être utilisé avec prudence chez les hommes fertiles. Dans des études chez la souris visant à évaluer l'évolution dans le temps et la réversibilité de la dégénérescence testiculaire induite par la ribavirine à des doses de 15 à 150 mg/kg/jour (équivalent humain estimé de 1,25 à 12,5 mg/kg/jour, sur la base d'un ajustement de la surface corporelle pour une adulte de 60 kg ; 0,1 à 0,8 fois la dose humaine maximale de ribavirine sur 24 heures) administrée pendant 3 ou 6 mois, des anomalies du sperme sont survenues. À l'arrêt du traitement, une récupération essentiellement totale de la toxicité testiculaire induite par la ribavirine était apparente en 1 ou 2 cycles de spermatogenèse.
Utilisation dans des populations spécifiques
Grossesse
Catégorie de grossesse X
[Voir CONTRE-INDICATIONS , AVERTISSEMENTS ET PRECAUTIONS , et Toxicologie non clinique ].
Traitement et post-traitement :
Risque potentiel pour le fœtus
La ribavirine est connue pour s'accumuler dans les composants intracellulaires d'où elle est éliminée très lentement. On ne sait pas si la ribavirine contenue dans le sperme exercera un effet tératogène potentiel lors de la fécondation des ovules. Dans une étude chez le rat, il a été conclu que la létalité dominante n'était pas induite par la ribavirine à des doses allant jusqu'à 200 mg/kg pendant 5 jours (doses équivalentes humaines estimées de 7,14 à 28,6 mg/kg, sur la base d'un ajustement de la surface corporelle pour une période de 60 kg adulte ; jusqu'à 1,7 fois la dose maximale recommandée de ribavirine chez l'homme). Cependant, en raison des effets tératogènes potentiels de la ribavirine chez l'homme, il convient de conseiller aux patients de sexe masculin de prendre toutes les précautions nécessaires pour éviter tout risque de grossesse pour leurs partenaires féminines.
Les femmes en âge de procréer ne doivent pas recevoir REBETOL à moins qu'elles n'utilisent une contraception efficace (deux formes fiables) pendant la période de traitement. De plus, une contraception efficace doit être utilisée pendant 6 mois après le traitement sur la base d'une demi-vie de doses multiples (t1/2) de ribavirine de 12 jours.
Les patients masculins et leurs partenaires féminines doivent pratiquer une contraception efficace (deux formes fiables) pendant le traitement par REBETOL et pendant la période post-thérapeutique de 6 mois (par exemple, 15 demi-vies pour la clairance de la ribavirine de l'organisme).
Un registre des grossesses à base de ribavirine a été établi pour surveiller les résultats materno-fœtaux des grossesses chez les patientes et les partenaires féminines de patients masculins exposés à la ribavirine pendant le traitement et pendant 6 mois après l'arrêt du traitement. Les médecins et les patients sont encouragés à signaler de tels cas en composant le 1-800-593-2214.
Mères allaitantes
On ne sait pas si le produit REBETOL est excrété dans le lait maternel. En raison du potentiel d'effets indésirables graves du médicament chez les nourrissons, une décision doit être prise soit d'arrêter l'allaitement, soit de retarder ou d'arrêter REBETOL.
Utilisation pédiatrique
La sécurité et l'efficacité de REBETOL 200 mg en association avec PegIntron n'ont pas été établies chez les patients pédiatriques de moins de 3 ans. Pour le traitement par REBETOL/INTRON A, les signes de progression de la maladie, tels que l'inflammation et la fibrose hépatiques, ainsi que les facteurs pronostiques de réponse, le génotype du VHC et la charge virale doivent être pris en compte lors de la décision de traiter un patient pédiatrique. Les avantages du traitement doivent être mis en balance avec les résultats d'innocuité observés.
Les données de suivi à long terme chez les sujets pédiatriques indiquent que REBETOL en association avec PegIntron ou avec INTRON A peut induire une inhibition de la croissance qui entraîne une taille réduite chez certains patients [voir AVERTISSEMENTS ET PRECAUTIONS et EFFETS INDÉSIRABLES ].
Les idées ou tentatives de suicide sont survenues plus fréquemment chez les patients pédiatriques, principalement les adolescents, par rapport aux patients adultes (2,4 % contre 1 %) pendant le traitement et le suivi hors traitement [voir AVERTISSEMENTS ET PRECAUTIONS ]. Comme chez les patients adultes, les patients pédiatriques ont présenté d'autres effets indésirables psychiatriques (p. ex., dépression, labilité émotionnelle, somnolence), anémie et neutropénie [voir AVERTISSEMENTS ET PRECAUTIONS ].
Utilisation gériatrique
Les essais cliniques de la thérapie REBETOL/INTRON A ou PegIntron n'ont pas inclus un nombre suffisant de sujets âgés de 65 ans et plus pour déterminer s'ils répondent différemment des sujets plus jeunes.
REBETOL 200 mg est connu pour être en grande partie excrété par les reins, et le risque de réactions toxiques à ce médicament peut être plus élevé chez les patients présentant une insuffisance rénale. Étant donné que les patients âgés ont souvent une fonction rénale diminuée, des précautions doivent être prises lors du choix de la dose. La fonction rénale doit être surveillée et des ajustements posologiques doivent être effectués en conséquence. REBETOL 200 mg ne doit pas être utilisé chez les patients dont la clairance de la créatinine est inférieure à 50 mL/min [voir CONTRE-INDICATIONS ].
En général, les gélules de REBETOL 200 mg doivent être administrées aux patients âgés avec prudence, en commençant par l'extrémité inférieure de la plage posologique, reflétant la plus grande fréquence de diminution de la fonction hépatique et cardiaque et de maladies concomitantes ou d'autres traitements médicamenteux. Dans les essais cliniques, les sujets âgés présentaient une fréquence d'anémie plus élevée (67 %) que les patients plus jeunes (28 %) [voir AVERTISSEMENTS ET PRECAUTIONS ].
Receveurs de greffes d'organes
L'innocuité et l'efficacité d'INTRON A et de PegIntron seuls ou en association avec REBETOL pour le traitement de l'hépatite C chez les receveurs de greffe de foie ou d'autres organes n'ont pas été établies. Dans un petit (n = 16) cas d'expérience monocentrique et non contrôlé, l'insuffisance rénale chez les receveurs d'allogreffe rénale recevant une association d'interféron alpha et de ribavirine était plus fréquente que prévu d'après l'expérience antérieure du centre avec des receveurs d'allogreffe rénale ne recevant pas d'association thérapeutique. La relation entre l'insuffisance rénale et le rejet de l'allogreffe rénale n'est pas claire.
Co-infection VIH ou VHB
La sécurité et l'efficacité de PegIntron/REBETOL 200 mg et INTRON A/REBETOL pour le traitement des patients atteints du VHC co-infectés par le VIH ou le VHB n'ont pas été établies.
SURDOSAGE
L'expérience en matière de surdosage est limitée. L'ingestion aiguë de jusqu'à 20 g de capsules REBETOL, l'ingestion d'INTRON A de jusqu'à 120 millions d'unités et les doses sous-cutanées d'INTRON A jusqu'à 10 fois les doses recommandées ont été annoncées. Les effets primaires qui ont été observés sont une augmentation de l'incidence et de la sévérité des effets indésirables liés à l'utilisation thérapeutique d'INTRON A et de REBETOL. Cependant, des anomalies des enzymes hépatiques, une insuffisance rénale, une hémorragie et un infarctus du myocarde ont été signalés lors de l'administration de doses sous-cutanées uniques d'INTRON A dépassant les recommandations posologiques.
Il n'y a pas d'antidote spécifique pour le surdosage d'INTRON A ou de REBETOL 200 mg, et l'hémodialyse et la dialyse péritonéale ne sont pas efficaces pour le traitement du surdosage de ces agents.
CONTRE-INDICATIONS
Le traitement combiné REBETOL est contre-indiqué dans :
- les femmes qui sont enceintes. REBETOL 200 mg peut nuire au fœtus lorsqu'il est administré à une femme enceinte. REBETOL est contre-indiqué chez les femmes enceintes ou susceptibles de l'être. Si REBETOL est utilisé pendant la grossesse, ou si la patiente tombe enceinte pendant qu'elle prend REBETOL 200 mg, la patiente doit être informée du danger potentiel pour son fœtus [voir AVERTISSEMENTS ET PRECAUTIONS , Utilisation dans des populations spécifiques , et INFORMATIONS PATIENTS ]
- hommes dont la partenaire féminine est enceinte
- les patients présentant des réactions d'hypersensibilité connues telles que le syndrome de Stevens-Johnson, une nécrolyse épidermique toxique et un érythème polymorphe à la ribavirine ou à tout composant du produit
- patients atteints d'hépatite auto-immune
- patients atteints d'hémoglobinopathies (p. ex., thalassémie majeure, anémie falciforme)
- patients dont la clairance de la créatinine est inférieure à 50 ml/min. [voir Utilisation dans des populations spécifiques et PHARMACOLOGIE CLINIQUE ]
- L'administration concomitante de REBETOL et de didanosine est contre-indiquée car l'exposition au métabolite actif de la didanosine (didésoxyadénosine 5'-triphosphate) est augmentée. Une insuffisance hépatique mortelle, ainsi qu'une neuropathie périphérique, une pancréatite et une hyperlactatémie/acidose lactique symptomatique ont été rapportées chez des patients recevant de la didanosine en association avec la ribavirine [voir INTERACTIONS MÉDICAMENTEUSES ].
PHARMACOLOGIE CLINIQUE
Mécanisme d'action
La ribavirine est un agent antiviral [voir Microbiologie ].
Pharmacocinétique
Les propriétés pharmacocinétiques des doses uniques et multiples chez l'adulte sont résumées dans le tableau 11. La ribavirine a été rapidement et largement absorbée après administration orale. Cependant, en raison du métabolisme de premier passage, la biodisponibilité absolue était en moyenne de 64 % (44 %). Il y avait une relation linéaire entre la dose et l'ASCtf (ASC du temps zéro à la dernière concentration mesurable) après des doses uniques de 200 à 1200 mg de ribavirine. La relation entre la dose et la Cmax était curviligne, tendant à devenir asymptote au-dessus de doses uniques de 400 à 600 mg.
Lors d'administrations orales multiples, sur la base de l'ASC12h, une accumulation de ribavirine multipliée par 6 a été observée dans le plasma. Suite à l'administration orale de 600 mg deux fois par jour, l'état d'équilibre a été atteint après environ 4 semaines, avec des concentrations plasmatiques moyennes à l'état d'équilibre de 2200 ng/mL (37 %). À l'arrêt du traitement, la demi-vie moyenne était de 298 (30 %) heures, ce qui reflète probablement une élimination lente des compartiments non plasmatiques.
Effet de l'antiacide sur l'absorption de la ribavirine
L'administration concomitante de capsules REBETOL avec un antiacide contenant du magnésium, de l'aluminium et de la siméthicone a entraîné une diminution de 14 % de l'ASCtf moyenne de la ribavirine. La pertinence clinique des résultats de cette étude à dose unique n'est pas connue.
Répartition tissulaire
Le transport de la ribavirine dans les compartiments non plasmatiques a été le plus largement étudié dans les globules rouges et a été identifié comme étant principalement via un transporteur de nucléosides équilibrant de type es. Ce type de transporteur est présent sur pratiquement tous les types de cellules et peut expliquer le volume de distribution important. La ribavirine ne se lie pas aux protéines plasmatiques.
Métabolisme et excrétion
La ribavirine a deux voies de métabolisme : (i) une voie de phosphorylation réversible dans les cellules nucléées ; et (ii) une voie de dégradation impliquant une déribosylation et une hydrolyse d'amide pour produire un métabolite d'acide triazole carboxylique. La ribavirine et ses métabolites triazole carboxamide et acide triazole carboxylique sont excrétés par les reins. Après administration orale de 600 mg de 14C-ribavirine, environ 61 % et 12 % de la radioactivité ont été éliminés dans l'urine et les fèces, respectivement, en 336 heures. La ribavirine inchangée représentait 17 % de la dose administrée.
Populations particulières
Dysfonctionnement rénal
La pharmacocinétique de la ribavirine a été évaluée après administration d'une dose orale unique (400 mg) de ribavirine à des sujets non infectés par le VHC présentant divers degrés d'insuffisance rénale. La valeur moyenne de l'ASCtf était trois fois plus élevée chez les sujets dont la clairance de la créatinine était comprise entre 10 et 30 ml/min par rapport aux sujets témoins (clairance de la créatinine supérieure à 90 ml/min). Chez les sujets dont les valeurs de clairance de la créatinine se situaient entre 30 et 60 ml/min, l'ASCtf était deux fois supérieure à celle des sujets témoins. L'augmentation de l'ASCtf semble être due à la réduction de la clairance rénale et non rénale chez ces sujets. Les essais d'efficacité de phase 3 ont inclus des sujets avec des valeurs de clairance de la créatinine supérieures à 50 mL/min. La pharmacocinétique de doses multiples de ribavirine ne peut pas être prédite avec précision chez les patients présentant un dysfonctionnement rénal. La ribavirine n'est pas éliminée efficacement par hémodialyse.
Les patients dont la clairance de la créatinine est inférieure à 50 mL/min ne doivent pas être traités par REBETOL [voir CONTRE-INDICATIONS ].
Dysfonctionnement hépatique
L'effet de la dysfonction hépatique a été évalué après une dose orale unique de ribavirine (600 mg). Les valeurs moyennes de l'ASCtf n'étaient pas significativement différentes chez les sujets présentant un dysfonctionnement hépatique léger, modéré ou sévère (Classification de Child-Pugh A, B ou C) par rapport aux sujets témoins. Cependant, les valeurs moyennes de la Cmax augmentaient avec la sévérité de la dysfonction hépatique et étaient deux fois plus élevées chez les sujets présentant une dysfonction hépatique sévère par rapport aux sujets témoins.
Patients âgés
Les évaluations pharmacocinétiques chez les sujets âgés n'ont pas été réalisées.
Le genre
Aucune différence pharmacocinétique cliniquement significative n'a été notée dans un essai à dose unique portant sur 18 sujets masculins et 18 sujets féminins.
Patients pédiatriques
Les propriétés pharmacocinétiques à doses multiples de REBETOL 200 mg gélules et d'INTRON A chez les enfants atteints d'hépatite C chronique âgés de 5 à 16 ans sont résumées dans le tableau 12. La pharmacocinétique de REBETOL 200 mg et d'INTRON A (dose normalisée) est similaire chez l'adulte et sujets pédiatriques.
Les caractéristiques pharmacocinétiques complètes de la solution buvable REBETOL n'ont pas été déterminées chez les sujets pédiatriques. Les valeurs de la Cmin de la ribavirine étaient similaires après l'administration de solution buvable REBETOL ou de capsules REBETOL 200 mg pendant 48 semaines de traitement chez des sujets pédiatriques (âgés de 3 à 16 ans).
Un essai clinique chez des sujets pédiatriques atteints d'hépatite C chronique âgés de 3 à 17 ans a été mené dans lequel la pharmacocinétique de PegIntron et REBETOL (gélules et solution buvable) a été évaluée. Chez les sujets pédiatriques recevant une dose ajustée en fonction de la surface corporelle de PegIntron à 60 mcg/m²/semaine, l'estimation du rapport logarithmique transformé de l'exposition pendant l'intervalle de dosage a été prédite comme étant de 58 % [IC à 90 % : 141 %, 177 %] supérieure à observée chez les adultes recevant 1,5 mcg/kg/semaine. La pharmacocinétique de REBETOL (dose normalisée) dans cet essai était similaire à celle rapportée dans une étude antérieure de REBETOL 200 mg en association avec INTRON A chez des sujets pédiatriques et chez des adultes.
Effet de la nourriture sur l'absorption de la ribavirine
L'ASCtf et la Cmax ont toutes deux augmenté de 70 % lorsque les gélules de REBETOL ont été administrées avec un repas riche en graisses (841 kcal, 53,8 g de matières grasses, 31,6 g de protéines et 57,4 g de glucides) dans une étude pharmacocinétique à dose unique [voir DOSAGE ET ADMINISTRATION ].
Microbiologie
Mécanisme d'action
Le mécanisme par lequel la ribavirine contribue à son efficacité antivirale en clinique n'est pas entièrement compris. La ribavirine a une activité antivirale directe en culture tissulaire contre de nombreux virus à ARN. La ribavirine augmente la fréquence des mutations dans les génomes de plusieurs virus et la ribavirine triphosphate inhibe la polymérase du VHC dans une réaction biochimique.
Activité antivirale en culture cellulaire
L'activité antivirale de la ribavirine dans le réplicon du VHC n'est pas bien comprise et n'a pas été définie en raison de la toxicité cellulaire de la ribavirine. Une activité antivirale directe a été observée dans la culture tissulaire d'autres virus à ARN. L'activité anti-VHC de l'interféron a été démontrée dans des cellules contenant des VHC-RNS auto-réplicatifs (cellules réplicons du VHC) ou une infection par le VHC.
La résistance
Les génotypes du VHC présentent une grande variabilité dans leur réponse au traitement par interféron humain recombinant pégylé/ribavirine. Les changements génétiques associés à la réponse variable n'ont pas été identifiés.
Résistance croisée
Il n'y a pas de résistance croisée rapportée entre les interférons pégylés/non pégylés et la ribavirine.
Toxicologie et pharmacologie animales
Etudes à long terme chez la souris et le rat [18 à 24 mois ; doses de 20 à 75 et 10 à 40 mg/kg/jour, respectivement (doses équivalentes humaines estimées de 1,67 à 6,25 et 1,43 à 5,71 mg/kg/jour, respectivement, basées sur l'ajustement de la surface corporelle pour un adulte de 60 kg ; environ 0,1 à 0,4 fois la dose maximale humaine de ribavirine sur 24 heures)] ont démontré une relation entre l'exposition chronique à la ribavirine et l'incidence accrue de lésions vasculaires (hémorragies microscopiques) chez la souris. Chez les rats, une dégénérescence rétinienne s'est produite chez les témoins, mais l'incidence a augmenté chez les rats traités à la ribavirine.
Dans une étude au cours de laquelle des ratons ont reçu une dose postnatale de ribavirine à des doses de 10, 25 et 50 mg/kg/jour, des décès liés au médicament sont survenus à 50 mg/kg (à des concentrations plasmatiques de ratons inférieures aux concentrations plasmatiques humaines chez l'humain). dose thérapeutique) entre les jours 13 et 48 de l'étude. Les ratons traités du 7e au 63e jour postnatal ont présenté une diminution mineure, liée à la dose, de la croissance globale à toutes les doses, qui s'est ensuite manifestée par de légères diminutions du poids corporel, de la longueur couronne-croupe, et la longueur osseuse. Ces effets ont montré des signes de réversibilité et aucun effet histopathologique sur les os n'a été observé. Aucun effet de la ribavirine n'a été observé sur le développement neurocomportemental ou reproductif.
Etudes cliniques
L'étude clinique 1 a évalué la monothérapie par PegIntron. Voir Étiquetage PegIntron pour plus d'informations sur cet essai.
Thérapie combinée REBETOL/PegIntron
Sujets adultes
Étude 2
Un essai randomisé a comparé le traitement avec deux régimes PegIntron/REBETOL de 200 mg [PegIntron 1,5 mcg/kg par voie sous-cutanée une fois par semaine/REBETOL 800 mg par voie orale par jour (en doses fractionnées) ; PegIntron 1,5 mcg/kg en sous-cutané une fois par semaine pendant 4 semaines puis 0,5 mcg/kg en sous-cutané une fois par semaine pendant 44 semaines/REBETOL 1000 ou 1200 mg par voie orale par jour (en doses fractionnées)] avec INTRON A [3 MUI en sous-cutané trois fois par semaine/REBETOL 1000 ou 1200 mg par voie orale par jour (en doses fractionnées)] chez 1530 adultes atteints d'hépatite C chronique. Les sujets n'ayant jamais reçu d'interféron ont été traités pendant 48 semaines et suivis pendant 24 semaines après le traitement. Les sujets éligibles avaient une maladie hépatique compensée, un ARN-VHC détectable, une ALT élevée et une histopathologie hépatique compatible avec une hépatite chronique.
La réponse au traitement a été définie comme un ARN-VHC indétectable 24 semaines après le traitement (voir Tableau 13). Le taux de réponse à la dose de PegIntron 1,5 mcg/kg et de ribavirine 800 mg était supérieur au taux de réponse à INTRON A/REBETOL (voir tableau 13). Le taux de réponse à PegIntron 1,5 → 0,5 mcg/kg/REBETOL 200 mg était essentiellement le même comme réponse à INTRON A/REBETOL (données non présentées).
Les sujets avec le génotype viral 1, quelle que soit leur charge virale, ont eu un taux de réponse inférieur à PegIntron (1,5 mcg/kg)/REBETOL (800 mg) par rapport aux sujets avec d'autres génotypes viraux. Les sujets présentant les deux facteurs de mauvais pronostic (génotype 1 et charge virale élevée) ont eu un taux de réponse de 30 % (78/256) par rapport à un taux de réponse de 29 % (71/247) avec la thérapie combinée INTRON A/REBETOL.
Les sujets ayant un poids corporel inférieur avaient tendance à avoir des taux d'effets indésirables plus élevés [voir EFFETS INDÉSIRABLES et des taux de réponse plus élevés que les sujets ayant un poids corporel plus élevé. Les différences dans les taux de réponse entre les bras de traitement n'ont pas sensiblement varié avec le poids corporel.
Les taux de réponse au traitement avec la thérapie combinée PegIntron/REBETOL étaient de 49 % chez les hommes et de 56 % chez les femmes. Les taux de réponse étaient plus faibles chez les sujets afro-américains et hispaniques et plus élevés chez les Asiatiques que chez les Caucasiens. Bien que les Afro-Américains aient eu une proportion plus élevée de facteurs de mauvais pronostic que les Caucasiens, le nombre de non-Caucasiens étudiés (11 % du total) était insuffisant pour permettre des conclusions significatives sur les différences de taux de réponse après ajustement des facteurs de pronostic dans cet essai.
Des biopsies hépatiques ont été obtenues avant et après le traitement chez 68 % des sujets. Par rapport aux valeurs initiales, environ les deux tiers des sujets de tous les groupes de traitement ont présenté une légère réduction de l'inflammation.
Étude 3
Dans un vaste essai communautaire aux États-Unis, 4913 sujets atteints d'hépatite C chronique ont été randomisés pour recevoir PegIntron 1,5 mcg/kg par voie sous-cutanée une fois par semaine en association avec une dose de REBETOL 200 mg de 800 à 1 400 mg (dosage basé sur le poids [WBD]) ou 800 mg (plat) par voie orale par jour (en doses fractionnées) pendant 24 ou 48 semaines en fonction du génotype. La réponse au traitement a été définie comme un ARN-VHC indétectable (sur la base d'un test avec une limite inférieure de détection de 125 UI/mL) à 24 semaines après le traitement.
Le traitement avec PegIntron 1,5 mcg/kg et REBETOL 800 à 1400 mg a entraîné une réponse virologique soutenue plus élevée par rapport à PegIntron en association avec une dose quotidienne plate de 800 mg de REBETOL. Les sujets pesant plus de 105 kg ont obtenu le plus grand bénéfice avec WBD, bien qu'un bénéfice modeste ait également été observé chez les sujets pesant plus de 85 à 105 kg (voir tableau 14). Le bénéfice de la WBD chez les sujets pesant plus de 85 kg a été observé avec les génotypes 1-3 du VHC. Les données disponibles étaient insuffisantes pour tirer des conclusions concernant d'autres génotypes. L'utilisation de WBD a entraîné une augmentation de l'incidence de l'anémie [voir EFFETS INDÉSIRABLES ].
Un total de 1552 sujets pesant plus de 65 kg dans l'étude 3 avaient le génotype 2 ou 3 et ont été randomisés pour 24 ou 48 semaines de traitement. Aucun bénéfice supplémentaire n'a été observé avec la durée de traitement plus longue.
Étude 4
Un grand essai randomisé a comparé l'innocuité et l'efficacité du traitement pendant 48 semaines avec deux régimes PegIntron/REBETOL 200 mg [PegIntron 1,5 mcg/kg et 1 mcg/kg par voie sous-cutanée une fois par semaine, tous deux en association avec REBETOL 800 à 1400 mg PO par jour (en deux doses)] et Pegasys 180 mcg par voie sous-cutanée une fois par semaine en association avec Copegus 1000 à 1200 mg PO par jour (en deux doses fractionnées) chez 3070 adultes naïfs de traitement atteints d'hépatite C chronique de génotype 1. Dans cet essai, l'absence de réponse virologique précoce (indétectable ARN-VHC ou réduction supérieure ou égale à 2 log10 par rapport à l'inclusion) par traitement La semaine 12 était le critère d'arrêt du traitement. La RVS a été définie comme un ARN-VHC indétectable (test Roche COBAS TaqMan, une limite inférieure de quantification de 27 UI/mL) à 24 semaines après le traitement (voir Tableau 15).
Les taux globaux de RVS étaient similaires parmi les trois groupes de traitement. Quel que soit le groupe de traitement, les taux de RVS étaient plus faibles chez les sujets présentant de mauvais facteurs pronostiques. Les sujets avec de mauvais facteurs pronostiques randomisés pour PegIntron (1,5 mcg/kg)/REBETOL ou Pegasys/Copegus, cependant, ont obtenu des taux de RVS plus élevés par rapport aux sujets similaires randomisés pour PegIntron 1 mcg/kg/REBETOL. Pour le PegIntron 1,5 mcg/kg et la dose de REBETOL, les taux de RVS pour les sujets avec et sans les facteurs pronostiques suivants étaient les suivants : cirrhose (10 % contre 42 %), taux d'ALT normaux (32 % contre 42 %), virus de base charge supérieure à 600 000 UI/mL (35 % contre 61 %), 40 ans et plus (38 % contre 50 %) et race afro-américaine (23 % contre 44 %). Chez les sujets avec un ARN-VHC indétectable à la semaine de traitement 12 qui ont reçu PegIntron (1,5 mcg/kg)/REBETOL 200 mg, le taux de RVS était de 81 % (328/407).
Étude 5 - Thérapie combinée REBETOL/PegIntron dans les échecs de traitement antérieurs
Dans un essai non comparatif, 2 293 sujets atteints de fibrose modérée à sévère en échec d'un traitement antérieur par l'association interféron alpha/ribavirine ont été retraités par PegIntron, 1,5 mcg/kg par voie sous-cutanée, une fois par semaine, en association avec de la ribavirine ajustée en fonction du poids. Les sujets éligibles comprenaient des non-répondeurs antérieurs (sujets qui étaient positifs pour l'ARN-VHC à la fin d'un minimum de 12 semaines de traitement) et des rechuteurs antérieurs (sujets qui étaient négatifs pour l'ARN-HCV à la fin d'un minimum de 12 semaines de traitement et qui ont ensuite rechuté après le post-traitement suivre). Les sujets négatifs à la semaine 12 ont été traités pendant 48 semaines et suivis pendant 24 semaines après le traitement. La réponse au traitement a été définie comme un ARN-VHC indétectable 24 semaines après le traitement (mesurée à l'aide d'un test basé sur la recherche, limite de détection de 125 UI/mL). Le taux de réponse global était de 22 % (497/2293) (IC à 99 % : 19,5, 23,9). Les sujets présentant les caractéristiques suivantes étaient moins susceptibles de bénéficier d'un retraitement : non-réponse antérieure, traitement antérieur par l'interféron pégylé, fibrose ou cirrhose en pont significative et infection de génotype 1.
Les taux de réponse virologique soutenue au retraitement selon les caractéristiques de base sont résumés dans le tableau 16.
L'obtention d'un ARN-VHC indétectable à la semaine de traitement 12 était un facteur prédictif puissant de RVS. Dans cet essai, 1 470 (64 %) sujets n'ont pas obtenu d'ARN-VHC indétectable à la 12e semaine de traitement, et on leur a proposé de participer à des essais de traitement à long terme, en raison d'une réponse inadéquate au traitement. Parmi les 823 (36 %) sujets dont l'ARN-VHC était indétectable à la semaine 12 du traitement, ceux infectés par le génotype 1 avaient une RVS de 48 % (245/507), avec une gamme de réponses par scores de fibrose (F4-F2) de 39-55 %. Les sujets infectés par le génotype 2/3 dont l'ARN du VHC était indétectable à la semaine 12 du traitement avaient une RVS globale de 70 % (196/281), avec une plage de réponses par scores de fibrose (F4-F2) de 60 à 83 %. Pour tous les génotypes, des scores de fibrose plus élevés étaient associés à une probabilité réduite d'obtenir une RVS.
Sujets pédiatriques
Les sujets pédiatriques âgés de 3 à 17 ans non traités auparavant avec une hépatite C chronique compensée et un ARN-VHC détectable ont été traités avec REBETOL 15 mg/kg par jour et PegIntron 60 mcg/m² une fois par semaine pendant 24 ou 48 semaines en fonction du génotype du VHC et de la charge virale de base. charger. Tous les sujets devaient être suivis pendant 24 semaines après le traitement. Un total de 107 sujets ont reçu un traitement, dont 52 % étaient des femmes, 89 % étaient de race blanche et 67 % étaient infectés par le génotype 1 du VHC. Sujets infectés par les génotypes 1, 4 ou le génotype 3 avec un ARN du VHC supérieur ou égal à 600 000 UI/mL ont reçu 48 semaines de traitement tandis que les personnes infectées par le génotype 2 ou le génotype 3 avec un ARN du VHC inférieur à 600 000 UI/mL ont reçu 24 semaines de traitement. Les résultats des essais sont résumés dans le tableau 17.
REBETOL/INTRON A Thérapie combinée
Sujets adultes
Sujets précédemment non traités
Des adultes atteints d'hépatite C chronique compensée et d'ARN-VHC détectable (évalué par un laboratoire central à l'aide d'un test RT-PCR basé sur la recherche) qui n'avaient pas été traités auparavant par un traitement par interféron alpha ont été inscrits à deux essais multicentriques en double aveugle (américains et internationaux) et randomisés pour recevoir REBETOL 200 mg gélules 1200 mg/jour (1000 mg/jour pour les sujets pesant moins ou égal à 75 kg) et INTRON A 3 MUI trois fois par semaine ou INTRON A et placebo pendant 24 ou 48 semaines suivies de 24 semaines de suivi hors thérapie. L'essai international ne comprenait pas de bras de traitement INTRON A et placebo de 24 semaines. L'essai américain a recruté 912 sujets qui, au départ, étaient 67 % d'hommes, 89 % de race blanche avec un score moyen de Knodell HAI (I+II+III) de 7,5 et 72 % de génotype 1. L'essai international, mené en Europe, en Israël , au Canada et en Australie, ont recruté 799 sujets (65 % d'hommes, 95 % de race blanche, score moyen de Knodell de 6,8 et 58 % de génotype 1).
Les résultats des essais sont résumés dans le tableau 18.
Parmi les sujets qui n'avaient pas atteint l'ARN du VHC en dessous de la limite de détection du test basé sur la recherche à la semaine 24 du traitement REBETOL / INTRON A, moins de 5% ont répondu à 24 semaines supplémentaires de traitement combiné.
Parmi les sujets atteints du génotype 1 du VHC traités avec la thérapie REBETOL/INTRON A qui ont atteint l'ARN du VHC en dessous de la limite de détection du test basé sur la recherche à 24 semaines, ceux randomisés à 48 semaines de traitement ont eu des réponses virologiques plus élevées que ceux du groupe 24- groupe de traitement d'une semaine. Il n'y avait aucune augmentation observée dans les taux de réponse pour les sujets avec HCV non-genotype 1 randomisés à la thérapie REBETOL/INTRON A depuis 48 semaines comparées à 24 semaines.
Sujets en rechute
Les sujets atteints d'hépatite C chronique compensée et d'ARN-VHC détectable (évalué par un laboratoire central à l'aide d'un test RT-PCR basé sur la recherche) qui avaient rechuté après un ou deux cycles de traitement par interféron (définis comme des taux sériques anormaux d'ALT) ont été inscrits dans deux essais multicentriques en double aveugle (américains et internationaux) et randomisés pour recevoir REBETOL 1200 mg/jour (1000 mg/jour pour les sujets pesant ≤ 75 kg) et INTRON A 3 MUI trois fois par semaine ou INTRON A et placebo pendant 24 semaines suivi de 24 semaines de suivi hors thérapie. L'essai américain a recruté 153 sujets qui, au départ, étaient 67 % d'hommes, 92 % de race blanche avec un score moyen de Knodell HAI (I+II+III) de 6,8 et 58 % de génotype 1. L'essai international, mené en Europe, en Israël , au Canada et en Australie, ont recruté 192 sujets (64 % d'hommes, 95 % de race blanche, score moyen de Knodell de 6,6 et 56 % de génotype 1). Les résultats des essais sont résumés dans le tableau 19.
Les réponses virologiques et histologiques étaient similaires chez les sujets masculins et féminins dans les essais non traités précédemment et dans les essais de rechute.
Sujets pédiatriques
Des sujets pédiatriques âgés de 3 à 16 ans atteints d'hépatite C chronique compensée et d'ARN-VHC détectable (évalué par un laboratoire central à l'aide d'un test RT-PCR basé sur la recherche) ont été traités avec REBETOL 15 mg/kg par jour et INTRON A 3 MUI/ m² trois fois par semaine pendant 48 semaines suivies de 24 semaines de suivi hors thérapie. Un total de 118 sujets ont reçu un traitement, dont 57 % étaient des hommes, 80 % de race blanche et 78 % de génotype 1. Les sujets de moins de 5 ans ont reçu la solution buvable REBETOL et ceux de 5 ans ou plus ont reçu soit la solution buvable REBETOL soit gélules.
Les résultats des essais sont résumés dans le tableau 20.
Les sujets avec le génotype viral 1, indépendamment de la charge virale, avaient un taux de réponse inférieur à la thérapie de combinaison d'INTRON A/REBETOL comparé aux sujets avec le génotype non-1, 36 % contre 81 %. Les sujets présentant les deux facteurs de mauvais pronostic (génotype 1 et charge virale élevée) avaient un taux de réponse de 26 % (13/50).
INFORMATIONS PATIENTS
Aucune information fournie. Veuillez vous référer au AVERTISSEMENTS ET PRECAUTIONS section.