Traitement pour une psychose: Strattera 10mg, 18mg, 25mg, 40mg Atomoxetine Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce que Strattera 40 mg et comment est-il utilisé ?
Strattera est un médicament d'ordonnance utilisé pour traiter les symptômes du trouble déficitaire de l'attention/hyperactivité (TDAH). Strattera 18 mg peut être utilisé seul ou avec d'autres médicaments.
Strattera 10 mg est un inhibiteur sélectif de la recapture de la noradrénaline.
On ne sait pas si Strattera est sûr et efficace chez les enfants de moins de 2 ans.
Quels sont les effets secondaires possibles de Strattera ?
Strattera 25 mg peut provoquer des effets indésirables graves, notamment :
- douleur thoracique,
- difficulté à respirer,
- étourdissement,
- hallucinations,
- de nouveaux problèmes de comportement,
- agression,
- hostilité,
- paranoïa,
- Douleur d'estomac,
- démangeaison,
- symptômes pseudo-grippaux,
- urine foncée,
- jaunisse (jaunissement de la peau ou des yeux),
- miction douloureuse ou difficile, et
- érection douloureuse ou qui dure plus de 4 heures
Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires les plus courants de Strattera 40 mg incluent :
- nausée,
- vomissement,
- maux d'estomac,
- constipation,
- bouche sèche,
- perte d'appétit,
- des changements d'humeur,
- se sentir fatigué,
- vertiges,
- problèmes de miction, et
- impuissance, difficulté à avoir une érection,
Dites au médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles de Strattera. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
ATTENTION
IDÉES SUICIDAIRES CHEZ LES ENFANTS ET LES ADOLESCENTS
STRATTERA (atomoxétine) a augmenté le risque d'idées suicidaires dans des études à court terme chez des enfants ou des adolescents atteints d'un trouble déficitaire de l'attention/hyperactivité (TDAH). Toute personne envisageant l'utilisation de STRATTERA 18 mg chez un enfant ou un adolescent doit équilibrer ce risque avec le besoin clinique. Les comorbidités associées au TDAH peuvent être associées à une augmentation du risque d'idées et/ou de comportements suicidaires. Les patients qui commencent un traitement doivent être étroitement surveillés afin de détecter toute tendance suicidaire (pensées et comportements suicidaires), une aggravation clinique ou des changements inhabituels de comportement. Les familles et les soignants doivent être informés de la nécessité d'une surveillance étroite et d'une communication avec le prescripteur. STRATTERA 10 mg est approuvé pour le TDAH chez les patients pédiatriques et adultes. STRATTERA 40 mg n'est pas approuvé pour le trouble dépressif majeur. Les analyses groupées d'essais contrôlés par placebo à court terme (6 à 18 semaines) de STRATTERA 10 mg chez les enfants et les adolescents (un total de 12 essais impliquant plus de 2200 patients, dont 11 essais dans le TDAH et 1 essai dans l'énurésie) ont révélé un risque plus élevé d'idées suicidaires au début du traitement chez les personnes recevant STRATTERA par rapport au placebo. Le risque moyen d'idées suicidaires chez les patients recevant STRATTERA 25 mg était de 0,4 % (5/1357 patients), contre aucun chez les patients sous placebo (851 patients). Aucun suicide n'est survenu dans ces essais [voir AVERTISSEMENTS ET PRECAUTIONS ].
LA DESCRIPTION
STRATTERA® (atomoxétine) est un inhibiteur sélectif du recaptage de la noradrénaline. L'atomoxétine HCl est l'isomère R(-) tel que déterminé par diffraction des rayons X. La désignation chimique est le chlorhydrate de (-)-N-méthyl-3-phényl-3-(o-tolyloxy)-propylamine. La formule moléculaire est C17H21NO•HCl, ce qui correspond à un poids moléculaire de 291,82. La structure chimique est :
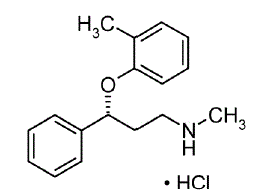
L'atomoxétine HCl est un solide blanc à pratiquement blanc, qui a une solubilité de 27,8 mg/mL dans l'eau.
Les gélules STRATTERA sont destinées à une administration orale uniquement.
Chaque gélule contient du chlorhydrate d'atomoxétine équivalent à 10, 18, 25, 40, 60, 80 ou 100 mg d'atomoxétine. Les gélules contiennent également de l'amidon prégélatinisé et de la diméthicone. Les enveloppes des gélules contiennent de la gélatine, du laurylsulfate de sodium et d'autres ingrédients inactifs. Les enveloppes des gélules contiennent également un ou plusieurs des éléments suivants :
FD&C Blue No. 2, oxyde de fer jaune synthétique, dioxyde de titane, oxyde de fer rouge. Les capsules sont imprimées avec de l'encre noire comestible.
LES INDICATIONS
Trouble déficitaire de l'attention/hyperactivité (TDAH)
STRATTERA 25 mg est indiqué pour le traitement du trouble déficitaire de l'attention/hyperactivité (TDAH).
L'efficacité des gélules STRATTERA a été établie dans sept essais cliniques chez des patients ambulatoires atteints de TDAH : quatre essais de 6 à 9 semaines chez des patients pédiatriques (âgés de 6 à 18 ans), deux essais de 10 semaines chez des adultes et un essai d'entretien en pédiatrie (âgés de 6 à 15) [voir Etudes cliniques ].
Considérations diagnostiques
Un diagnostic de TDAH (DSM-IV) implique la présence de symptômes hyperactifs-impulsifs ou inattentifs qui causent une déficience et qui étaient présents avant l'âge de 7 ans. Les symptômes doivent être persistants, doivent être plus graves que ceux généralement observés chez les individus à un niveau de développement comparable, doivent entraîner une altération cliniquement significative, par exemple, dans le fonctionnement social, scolaire ou professionnel, et doivent être présents dans 2 contextes ou plus, par exemple, à l'école (ou au travail) et à la maison. Les symptômes ne doivent pas être mieux expliqués par un autre trouble mental.
L'étiologie spécifique du TDAH est inconnue et il n'existe pas de test diagnostique unique. Un diagnostic adéquat nécessite l'utilisation non seulement de ressources médicales mais aussi de ressources psychologiques, éducatives et sociales spéciales. L'apprentissage peut être altéré ou non. Le diagnostic doit être basé sur une anamnèse complète et une évaluation du patient et non uniquement sur la présence du nombre requis de caractéristiques du DSM-IV.
Pour le type inattentif, au moins 6 des symptômes suivants doivent avoir persisté pendant au moins 6 mois : manque d'attention aux détails/erreurs d'inattention, manque d'attention soutenue, mauvaise écoute, incapacité à mener à bien les tâches, mauvaise organisation, évite les tâches exigeant un effort mental soutenu, perd des choses, facilement distrait, oublieux. Pour le type hyperactif-impulsif, au moins 6 des symptômes suivants doivent avoir persisté pendant au moins 6 mois : gigoter/se tortiller, quitter son siège, courir/escalader de manière inappropriée, difficulté à effectuer des activités calmes, "en déplacement", parler de manière excessive, bavardage réponses, ne peut pas attendre son tour, intrusif. Pour un diagnostic de type combiné, les critères inattentifs et hyperactifs-impulsifs doivent être remplis.
Besoin d'un programme de traitement complet
STRATTERA 18 mg est indiqué dans le cadre d'un programme de traitement global du TDAH pouvant inclure d'autres mesures (psychologiques, éducatives, sociales) pour les patients atteints de ce syndrome. Le traitement médicamenteux peut ne pas être indiqué pour tous les patients atteints de ce syndrome. Le traitement médicamenteux n'est pas destiné à être utilisé chez le patient qui présente des symptômes secondaires à des facteurs environnementaux et/ou à d'autres troubles psychiatriques primaires, y compris la psychose. Un placement éducatif approprié est essentiel chez les enfants et les adolescents avec ce diagnostic et une intervention psychosociale est souvent utile. Lorsque les mesures correctives seules sont insuffisantes, la décision de prescrire un traitement médicamenteux dépendra de l'évaluation par le médecin de la chronicité et de la gravité des symptômes du patient.
DOSAGE ET ADMINISTRATION
Traitement aigu
Dosage des enfants et des adolescents jusqu'à 70 kg de poids corporel
STRATTERA 40 mg doit être initié à une dose quotidienne totale d'environ 0,5 mg/kg et augmentée après un minimum de 3 jours jusqu'à une dose quotidienne totale cible d'environ 1,2 mg/kg administrée soit en une dose quotidienne unique le matin, soit en doses égales. doses le matin et en fin d'après-midi/début de soirée. Aucun bénéfice supplémentaire n'a été démontré pour des doses supérieures à 1,2 mg/kg/jour [voir Etudes cliniques ].
La dose quotidienne totale chez les enfants et les adolescents ne doit pas dépasser 1,4 mg/kg ou 100 mg, selon la valeur la moins élevée.
Dosage des enfants et des adolescents de plus de 70 kg de poids corporel et des adultes
STRATTERA 18 mg doit être initié à une dose quotidienne totale de 40 mg et augmentée après un minimum de 3 jours jusqu'à une dose quotidienne totale cible d'environ 80 mg administrée soit en une dose quotidienne unique le matin, soit en doses uniformément réparties le matin et fin d'après-midi/début de soirée. Après 2 à 4 semaines supplémentaires, la dose peut être augmentée jusqu'à un maximum de 100 mg chez les patients n'ayant pas obtenu une réponse optimale. Il n'y a pas de données qui soutiennent une efficacité accrue à des doses plus élevées [voir Etudes cliniques ].
La dose quotidienne totale maximale recommandée chez les enfants et les adolescents de plus de 70 kg et les adultes est de 100 mg.
Traitement d'entretien/prolongé
Il est généralement admis que le traitement pharmacologique du TDAH peut être nécessaire pendant de longues périodes. Le bénéfice de maintenir les patients pédiatriques (âgés de 6 à 15 ans) atteints de TDAH sous STRATTERA 18 mg après avoir obtenu une réponse dans une plage de doses de 1,2 à 1,8 mg/kg/jour a été démontré dans un essai contrôlé. Les patients assignés à STRATTERA dans la phase d'entretien ont généralement continué à recevoir la même dose utilisée pour obtenir une réponse dans la phase en ouvert. Le médecin qui choisit d'utiliser STRATTERA pendant des périodes prolongées doit réévaluer périodiquement l'utilité à long terme du médicament pour le patient individuel [voir Etudes cliniques ].
Informations générales sur le dosage
STRATTERA peut être pris avec ou sans nourriture.
STRATTERA 40mg peut être interrompu sans être dégressif.
Les gélules de STRATTERA ne sont pas destinées à être ouvertes, elles doivent être prises entières [voir INFORMATIONS PATIENTS ].
La sécurité des doses uniques supérieures à 120 mg et des doses quotidiennes totales supérieures à 150 mg n'a pas été systématiquement évaluée.
Dosage dans des populations spécifiques
Ajustement posologique pour les patients atteints d'insuffisance hépatique
Pour les patients atteints de TDAH qui présentent une insuffisance hépatique (IH), un ajustement posologique est recommandé comme suit : pour les patients atteints d'IH modérée (classe B de Child-Pugh), les doses initiales et cibles doivent être réduites à 50 % de la dose normale (pour les patients sans SALUT). Pour les patients atteints d'IH sévère (classe C de Child-Pugh), la dose initiale et les doses cibles doivent être réduites à 25 % de la normale [voir Utilisation dans des populations spécifiques ].
Ajustement de la posologie pour une utilisation avec un inhibiteur puissant du CYP2D6 ou chez les patients connus pour être des PM du CYP2D6
Chez les enfants et les adolescents pesant jusqu'à 70 kg recevant des inhibiteurs puissants du CYP2D6, par exemple la paroxétine, la fluoxétine et la quinidine, ou chez les patients connus pour être des PM du CYP2D6, STRATTERA doit être initié à 0,5 mg/kg/jour et augmenté uniquement jusqu'à la dose cible habituelle de 1,2 mg/kg/jour si les symptômes ne s'améliorent pas après 4 semaines et si la dose initiale est bien tolérée.
Chez les enfants et les adolescents de plus de 70 kg de poids corporel et les adultes recevant des inhibiteurs puissants du CYP2D6, par exemple la paroxétine, la fluoxétine et la quinidine, STRATTERA doit être initié à 40 mg/jour et uniquement augmenté à la dose cible habituelle de 80 mg/jour en cas d'échec des symptômes. s'améliore après 4 semaines et la dose initiale est bien tolérée.
COMMENT FOURNIE
Formes posologiques et points forts
Chaque gélule contient du chlorhydrate d'atomoxétine équivalent à 10 mg (blanc opaque, blanc opaque), 18 mg (or, blanc opaque), 25 mg (bleu opaque, blanc opaque), 40 mg (bleu opaque, bleu opaque), 60 mg (opaque Blue, Gold), 80 mg (Opaque Brown, Opaque White) ou 100 mg (Opaque Brown, Opaque Brown) d'atomoxétine.
Stockage et manutention
Conserver à 25 °C (77 °F); les excursions permises à 15° à 30°C (59° à 86°F) [voient USP la Température de Pièce Contrôlée].
Commercialisé par : Lilly USA, LLC Indianapolis, IN 46285, États-Unis. Révisé : mai 2017.
EFFETS SECONDAIRES
Expérience des essais cliniques
STRATTERA 10 mg a été administré à 5 382 enfants ou adolescents atteints de TDAH et à 1 007 adultes atteints de TDAH dans des études cliniques. Au cours des essais cliniques sur le TDAH, 1625 enfants et adolescents ont été traités pendant plus d'un an et 2529 enfants et adolescents ont été traités pendant plus de 6 mois.
Étant donné que les essais cliniques sont menés dans des conditions très variables, les taux d'effets indésirables observés dans les essais cliniques d'un médicament ne peuvent pas être directement comparés aux taux des essais cliniques d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique.
Essais cliniques sur les enfants et les adolescents
Raisons de l'arrêt du traitement en raison d'effets indésirables lors d'essais cliniques sur des enfants et des adolescents
Dans des essais contrôlés par placebo chez des enfants et des adolescents en phase aiguë, 3,0 % (48/1613) des sujets sous atomoxétine et 1,4 % (13/945) des sujets sous placebo ont arrêté le traitement en raison d'effets indésirables. Pour toutes les études (y compris les études ouvertes et à long terme), 6,3 % des patients métaboliseurs rapides (EM) et 11,2 % des patients métaboliseurs lents (MP) ont arrêté en raison d'un effet indésirable. Chez les patients traités par STRATTERA, irritabilité (0,3 %, N = 5) ; somnolence (0,3 %, N=5) ; agressivité (0,2 %, N = 4) ; nausées (0,2 %, N = 4) ; vomissements (0,2 %, N = 4 ); douleurs abdominales (0,2 %, N = 4) ; constipation (0,1 %, N=2) ; fatigue (0,1 %, N=2) ; sensation anormale (0,1 %, N = 2) ; et les céphalées (0,1 %, N = 2) étaient les raisons de l'arrêt signalées par plus d'un patient.
Saisies
STRATTERA n'a pas été systématiquement évalué chez les patients pédiatriques souffrant de troubles convulsifs, car ces patients ont été exclus des études cliniques lors des tests de précommercialisation du produit. Dans le programme de développement clinique, des convulsions ont été signalées chez 0,2 % (12/5073) des enfants dont l'âge moyen était de 10 ans (intervalle de 6 à 16 ans). Dans ces essais cliniques, le risque de convulsions chez les métaboliseurs lents était de 0,3 % (1/293) contre 0,2 % (11/4741) chez les métaboliseurs rapides.
Effets indésirables fréquemment observés dans les essais contrôlés par placebo sur des enfants et des adolescents en phase aiguë
Les effets indésirables couramment observés associés à l'utilisation de STRATTERA (incidence de 2 % ou plus) et non observés à une incidence équivalente chez les patients traités par placebo (incidence de STRATTERA 40 mg supérieure à celle du placebo) sont répertoriés dans le tableau 2. Les résultats étaient similaires dans l'étude BID et l'essai QD, sauf comme indiqué dans le tableau 3, qui montre les résultats BID et QD pour les effets indésirables sélectionnés sur la base de tests de Breslow-Day statistiquement significatifs. Les effets indésirables les plus fréquemment observés chez les patients traités par STRATTERA (incidence de 5 % ou plus et au moins le double de l'incidence chez les patients sous placebo, pour l'administration BID ou QD) étaient : nausées, vomissements, fatigue, diminution de l'appétit, douleurs abdominales et somnolence (voir Tableaux 2 et 3).
Des données supplémentaires provenant d'essais cliniques sur le TDAH (contrôlés et non contrôlés) ont montré qu'environ 5 à 10 % des patients pédiatriques ont présenté des modifications potentiellement importantes sur le plan clinique de la fréquence cardiaque (≥ 20 battements par minute) ou de la tension artérielle (≥ 15 à 20 mm Hg) [voir CONTRE-INDICATIONS et AVERTISSEMENTS ET PRECAUTIONS ].
Les effets indésirables suivants sont survenus chez au moins 2 % des patients enfants et adolescents PM CYP2D6 et étaient statistiquement significativement plus fréquents chez les patients PM que chez les patients EM CYP2D6 : insomnie (11 % des PM, 6 % des EM) ; poids diminué (7 % des PM, 4 % des EM) ; constipation (7 % des PM, 4 % des EM) ; dépression1 (7 % des PM, 4 % des EM) ; tremblement (5 % des PM, 1 % des EM) ; excoriation (4 % des PM, 2 % des EM) ; insomnie moyenne (3 % des PM, 1 % des EM) ; conjonctivite (3 % des PM, 1 % des EM) ; syncope (3 % des PM, 1 % des EM) ; réveil matinal (2 % des PM, 1 % des EM) ; mydriase (2 % des PM, 1 % des EM) ; sédation (4 % des PM, 2 % des EM).
1Dépression comprend les termes suivants : dépression, dépression majeure, symptômes dépressifs, humeur dépressive, dysphorie.
Essais cliniques adultes
Raisons de l'arrêt du traitement en raison d'effets indésirables lors d'études aiguës contrôlées par placebo chez l'adulte
Dans les procès contrôlés du placebo adultes aigus, 11.3 % (61/541) les sujets d'atomoxetine et 3.0 % (12/405) les sujets de placebo ont arrêté pour les réactions défavorables. Parmi les patients traités par STRATTERA, insomnie (0,9 %, N = 5) ; nausées (0,9 %, N = 5 ); douleur thoracique (0,6 %, N = 3) ; fatigue (0,6 %, N=3) ; anxiété (0,4 %, N=2) ; dysfonction érectile (0,4 %, N=2) ; sautes d'humeur (0,4 %, N = 2); nervosité (0,4 %, N=2) ; palpitations (0,4 %, N=2) ; et la rétention urinaire (0,4 %, N = 2) étaient les raisons de l'arrêt rapportées par plus d'un patient.
Saisies
STRATTERA n'a pas fait l'objet d'une évaluation systématique chez les patients adultes présentant un trouble convulsif car ces patients ont été exclus des études cliniques lors des tests de précommercialisation du produit. Dans le programme de développement clinique, des convulsions ont été signalées chez 0,1 % (1/748) des patients adultes. Dans ces essais cliniques, aucun métaboliseur lent (0/43) n'a signalé de convulsions contre 0,1 % (1/705) pour les métaboliseurs rapides.
Effets indésirables couramment observés dans les essais contrôlés par placebo chez l'adulte
Les effets indésirables fréquemment observés associés à l'utilisation de STRATTERA (incidence de 2 % ou plus) et non observés à une incidence équivalente chez les patients traités par placebo (incidence de STRATTERA 25 mg supérieure à celle du placebo) sont répertoriés dans le tableau 4. Les effets indésirables les plus fréquemment observés chez les patients traités par STRATTERA (incidence de 5 % ou plus et au moins le double de l'incidence chez les patients sous placebo) étaient : constipation, bouche sèche, nausées, diminution de l'appétit, étourdissements, dysfonction érectile et hésitation urinaire (voir le tableau 4). Des données supplémentaires provenant d'essais cliniques sur le TDAH (contrôlés et non contrôlés) ont montré qu'environ 5 à 10 % des patients adultes présentaient des modifications potentiellement importantes sur le plan clinique de la fréquence cardiaque (≥20 battements par minute) ou de la tension artérielle (≥15 à 20 mm Hg) [voir CONTRE-INDICATIONS et AVERTISSEMENTS ET PRECAUTIONS ].
Les événements indésirables suivants sont survenus chez au moins 2 % des patients adultes métaboliseurs lents (MP) du CYP2D6 et étaient statistiquement significativement plus fréquents chez les patients métaboliseurs lents (MP) du CYP2D6 que chez les patients métaboliseurs rapides (EM) du CYP2D6 : vision floue (4 % des PM, 1 % des EM ); sécheresse de la bouche (35 % des PM, 17 % des EM) ; constipation (11 % des PM, 7 % des EM) ; se sentir nerveux (5 % des PM, 2 % des EM) ; diminution de l'appétit (23 % des PM, 15 % des EM) ; tremblement (5 % des PM, 1 % des EM) ; insomnie (19 % des PM, 11 % des EM) ; trouble du sommeil (7 % des PM, 3 % des EM) ; insomnie moyenne (5 % des PM, 3 % des EM) ; insomnie terminale (3 % des PM, 1 % des EM) ; rétention urinaire (6 % des PM, 1 % des EM) ; dysfonction érectile (21 % des PM, 9 % des EM) ; trouble de l'éjaculation (6 % des PM, 2 % des EM) ; hyperhidrose (15 % des PM, 7 % des EM) ; froideur périphérique (3% des PM, 1% des EM).
Dysfonction sexuelle masculine et féminine
L'atomoxétine semble altérer la fonction sexuelle chez certains patients. Les modifications du désir sexuel, de la performance sexuelle et de la satisfaction sexuelle ne sont pas bien évaluées dans la plupart des essais cliniques car elles nécessitent une attention particulière et parce que les patients et les médecins peuvent être réticents à en discuter. En conséquence, les estimations de l'incidence des expériences et des performances sexuelles indésirables citées dans l'étiquetage des produits sous-estiment probablement l'incidence réelle. Le tableau 4 ci-dessus présente l'incidence des effets secondaires sexuels signalés par au moins 2 % des patients adultes prenant STRATTERA dans des essais contrôlés par placebo.
Il n'y a pas d'études adéquates et bien contrôlées examinant la dysfonction sexuelle avec le traitement par STRATTERA 40 mg. Bien qu'il soit difficile de connaître le risque précis de dysfonctionnement sexuel associé à l'utilisation de STRATTERA 40 mg, les médecins doivent systématiquement se renseigner sur ces éventuels effets secondaires.
Rapports spontanés post-commercialisation
Les effets indésirables suivants ont été identifiés lors de l'utilisation post-approbation de STRATTERA. Sauf indication contraire, ces effets indésirables sont survenus chez des adultes, des enfants et des adolescents. Étant donné que ces réactions sont signalées volontairement par une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale avec l'exposition au médicament.
Système cardiovasculaire - Allongement du QT, syncope.
Effets vasculaires périphériques - Phénomène de raynaud.
Troubles généraux et anomalies au site d'administration - Léthargie.
Système musculo-squelettique - Rhabdomyolyse.
Troubles du système nerveux - Hypoesthésie ; paresthésie chez les enfants et les adolescents ; troubles sensoriels; tics.
Troubles psychiatriques - Dépression et humeur dépressive ; anxiété, changements de libido.
Saisies Des convulsions ont été signalées au cours de la période suivant la commercialisation. Les cas de convulsions post-commercialisation incluent des patients présentant des troubles convulsifs préexistants et ceux présentant des facteurs de risque identifiés pour les convulsions, ainsi que des patients sans antécédent ni facteurs de risque identifiés pour les convulsions. La relation exacte entre STRATTERA 40 mg et les convulsions est difficile à évaluer en raison de l'incertitude quant au risque de fond de convulsions chez les patients atteints de TDAH.
Affections de la peau et du tissu sous-cutané - Alopécie, hyperhidrose.
Système urogénital - Douleur pelvienne masculine ; hésitation urinaire chez les enfants et les adolescents; rétention urinaire chez l'enfant et l'adolescent.
INTERACTIONS MÉDICAMENTEUSES
Inhibiteurs de la monoamine oxydase
Avec d'autres médicaments qui affectent les concentrations cérébrales de monoamines, des réactions graves, parfois mortelles, ont été signalées (notamment hyperthermie, rigidité, myoclonie, instabilité autonome avec possibles fluctuations rapides des signes vitaux et modifications de l'état mental, notamment une agitation extrême évoluant vers le délire et le coma ) lorsqu'il est pris en combinaison avec un IMAO. Certains cas présentaient des caractéristiques ressemblant au syndrome malin des neuroleptiques. De telles réactions peuvent survenir lorsque ces médicaments sont administrés simultanément ou à proximité [voir CONTRE-INDICATIONS ].
Effet des inhibiteurs du CYP2D6 sur l'atomoxétine
Chez les métaboliseurs rapides (EM), les inhibiteurs du CYP2D6 (par exemple, la paroxétine, la fluoxétine et la quinidine) augmentent les concentrations plasmatiques d'atomoxétine à l'état d'équilibre jusqu'à des expositions similaires à celles observées chez les métaboliseurs lents (MP). Chez les personnes EM traitées avec de la paroxétine ou de la fluoxétine, l'ASC de l'atomoxétine est d'environ 6 à 8 fois et la Css, max est d'environ 3 à 4 fois supérieure à celle de l'atomoxétine seule.
Des études in vitro suggèrent que la co-administration d'inhibiteurs du cytochrome P450 à des PM n'augmentera pas les concentrations plasmatiques d'atomoxétine.
Médicaments antihypertenseurs et agents vasopresseurs
En raison des effets possibles sur la tension artérielle, STRATTERA doit être utilisé avec prudence avec des médicaments antihypertenseurs et des agents vasopresseurs (p. ex., dopamine, dobutamine) ou d'autres médicaments qui augmentent la tension artérielle.
Albutérol
STRATTERA doit être administré avec prudence aux patients traités par l'albutérol administré par voie systémique (orale ou intraveineuse) (ou d'autres bêta2-agonistes) car l'action de l'albutérol sur le système cardiovasculaire peut être potentialisée, entraînant une augmentation de la fréquence cardiaque et de la pression artérielle. L'albutérol (600 mcg iv pendant 2 heures) a induit des augmentations de la fréquence cardiaque et de la tension artérielle. Ces effets ont été potentialisés par l'atomoxétine (60 mg bid pendant 5 jours) et ont été plus marqués après la co-administration initiale d'albutérol et d'atomoxétine. Cependant, ces effets sur la fréquence cardiaque et la pression artérielle n'ont pas été observés dans une autre étude après la co-administration avec une dose inhalée d'albutérol (200-800 mcg) et d'atomoxétine (80 mg QD pendant 5 jours) chez 21 sujets asiatiques en bonne santé qui ont été exclus pour de faibles statut de métaboliseur.
Effet de l'atomoxétine sur les enzymes P450
L'atomoxétine n'a pas provoqué d'inhibition ou d'induction cliniquement importante des enzymes du cytochrome P450, y compris CYP1A2, CYP3A, CYP2D6 et CYP2C9.
Substrat du CYP3A (p. ex., Midazolam)
L'administration concomitante de STRATTERA (60 mg BID pendant 12 jours) avec le midazolam, un composé modèle pour les médicaments métabolisés par le CYP3A4 (dose unique de 5 mg), a entraîné une augmentation de 15 % de l'ASC du midazolam. Aucun ajustement posologique n'est recommandé pour les médicaments métabolisés par le CYP3A.
Substrat du CYP2D6 (p. ex. désipramine)
L'administration concomitante de STRATTERA (40 ou 60 mg deux fois par jour pendant 13 jours) avec la désipramine, un composé modèle pour les médicaments métabolisés par le CYP2D6 (dose unique de 50 mg), n'a pas modifié la pharmacocinétique de la désipramine. Aucun ajustement posologique n'est recommandé pour les médicaments métabolisés par le CYP2D6.
De l'alcool
La consommation d'éthanol avec STRATTERA 18mg n'a pas modifié les effets intoxicants de l'éthanol.
Méthylphénidate
L'administration concomitante de méthylphénidate avec STRATTERA 18 mg n'a pas augmenté les effets cardiovasculaires au-delà de ceux observés avec le méthylphénidate seul.
Médicaments fortement liés aux protéines plasmatiques
Des études in vitro de déplacement de médicament ont été menées avec l'atomoxétine et d'autres médicaments hautement liés à des concentrations thérapeutiques. L'atomoxétine n'a pas affecté la liaison de la warfarine, de l'acide acétylsalicylique, de la phénytoïne ou du diazépam à l'albumine humaine. De même, ces composés n'ont pas affecté la liaison de l'atomoxétine à l'albumine humaine.
Médicaments qui affectent le pH gastrique
Les médicaments qui élèvent le pH gastrique (hydroxyde de magnésium/hydroxyde d'aluminium, oméprazole) n'ont eu aucun effet sur la biodisponibilité de STRATTERA.
Toxicomanie et dépendance
Substance contrôlée
STRATTERA n'est pas une substance contrôlée.
Abuser de
Dans une étude randomisée, en double aveugle, contrôlée par placebo et sur le potentiel d'abus chez des adultes comparant les effets de STRATTERA et d'un placebo, STRATTERA n'a pas été associé à un schéma de réponse suggérant des propriétés stimulantes ou euphorisantes.
Dépendance
Les données d'études cliniques portant sur plus de 2 000 enfants, adolescents et adultes atteints de TDAH et sur plus de 1 200 adultes souffrant de dépression n'ont montré que des incidents isolés de détournement de médicament ou d'auto-administration inappropriée associés à STRATTERA. Il n'y avait aucune preuve de rebond des symptômes ou d'effets indésirables suggérant un syndrome d'arrêt du traitement ou de sevrage.
Expérience animale
Les études de discrimination des drogues chez les rats et les singes ont montré une généralisation incohérente des stimuli entre l'atomoxétine et la cocaïne.
AVERTISSEMENTS
Inclus dans le cadre du "PRÉCAUTIONS" Section
PRÉCAUTIONS
Idéation suicidaire
STRATTERA a augmenté le risque d'idées suicidaires dans des études à court terme chez des enfants et des adolescents atteints d'un trouble déficitaire de l'attention/hyperactivité (TDAH). Les analyses groupées d'essais contrôlés par placebo à court terme (6 à 18 semaines) de STRATTERA chez les enfants et les adolescents ont révélé un risque accru d'idées suicidaires au début du traitement chez les personnes recevant STRATTERA. Il y avait un total de 12 essais (11 dans le TDAH et 1 dans l'énurésie) impliquant plus de 2200 patients (dont 1357 patients recevant STRATTERA et 851 recevant un placebo). Le risque moyen d'idées suicidaires chez les patients recevant STRATTERA était de 0,4 % (5/1357 patients), contre aucun chez les patients sous placebo. Il y a eu 1 tentative de suicide parmi ces environ 2200 patients, survenue chez un patient traité par STRATTERA. Aucun suicide n'est survenu dans ces essais. Toutes les réactions sont survenues chez des enfants de 12 ans ou moins. Toutes les réactions sont survenues au cours du premier mois de traitement. On ne sait pas si le risque d'idées suicidaires chez les patients pédiatriques s'étend à une utilisation à plus long terme. Une analyse similaire chez des patients adultes traités par STRATTERA pour un TDAH ou un trouble dépressif majeur (TDM) n'a pas révélé de risque accru d'idées ou de comportements suicidaires en association avec l'utilisation de STRATTERA.
Tous les patients pédiatriques traités par STRATTERA doivent être surveillés de manière appropriée et observés de près pour déceler une aggravation clinique, des tendances suicidaires et des changements de comportement inhabituels, en particulier au cours des premiers mois d'un traitement médicamenteux, ou lors de changements de dose, soit des augmentations ou des diminutions. .
Les symptômes suivants ont été signalés avec STRATTERA : anxiété, agitation, attaques de panique, insomnie, irritabilité, hostilité, agressivité, impulsivité, akathisie (agitation psychomotrice), hypomanie et manie. Bien qu'un lien de causalité entre l'émergence de tels symptômes et l'émergence d'impulsions suicidaires n'ait pas été établi, on craint que ces symptômes puissent représenter des précurseurs d'une suicidalité émergente. Ainsi, les patients traités par STRATTERA 40 mg doivent être surveillés pour l'apparition de tels symptômes.
Il convient d'envisager de modifier le schéma thérapeutique, y compris éventuellement d'arrêter le médicament, chez les patients qui présentent des tendances suicidaires émergentes ou des symptômes qui pourraient être des précurseurs de tendances suicidaires émergentes, en particulier si ces symptômes sont graves ou d'apparition brutale, ou ne faisaient pas partie du traitement. symptômes présentés par le patient.
Les familles et les soignants des patients pédiatriques traités par STRATTERA 10 mg doivent être avertis de la nécessité de surveiller l'apparition d'agitation, d'irritabilité, de changements inhabituels de comportement et des autres symptômes décrits ci-dessus, ainsi que l'apparition de tendances suicidaires, et de surveiller les patients. signalez immédiatement ces symptômes aux fournisseurs de soins de santé. Une telle surveillance devrait inclure une observation quotidienne par les familles et les soignants.
Lésions hépatiques graves
Les rapports post-commercialisation indiquent que STRATTERA peut causer de graves lésions hépatiques. Bien qu'aucune preuve de lésion hépatique n'ait été détectée dans les essais cliniques portant sur environ 6000 patients, il y a eu de rares cas de lésions hépatiques cliniquement significatives qui ont été considérées comme probablement ou possiblement liées à l'utilisation de STRATTERA 40 mg dans l'expérience post-commercialisation. De rares cas d'insuffisance hépatique ont également été signalés, dont un cas ayant entraîné une greffe du foie. En raison d'une sous-déclaration probable, il est impossible de fournir une estimation précise de l'incidence réelle de ces réactions. Les cas signalés de lésions hépatiques sont survenus dans les 120 jours suivant le début de l'atomoxétine dans la majorité des cas et certains patients présentaient des enzymes hépatiques nettement élevées [> 20 X la limite supérieure de la normale (LSN)] et un ictère avec des taux de bilirubine significativement élevés (> 2 X LSN), suivie d'une récupération à l'arrêt de l'atomoxétine. Chez un patient, une lésion hépatique, se manifestant par une élévation des enzymes hépatiques jusqu'à 40 X LSN et un ictère avec bilirubine jusqu'à 12 X LSN, est réapparue lors d'une nouvelle provocation et a été suivie d'un rétablissement à l'arrêt du médicament, ce qui prouve que STRATTERA a probablement causé la lésion hépatique. De telles réactions peuvent survenir plusieurs mois après le début du traitement, mais les anomalies biologiques peuvent continuer à s'aggraver pendant plusieurs semaines après l'arrêt du médicament. Le patient décrit ci-dessus s'est rétabli de sa lésion hépatique et n'a pas eu besoin d'une greffe de foie.
STRATTERA doit être arrêté chez les patients présentant un ictère ou des signes biologiques de lésions hépatiques, et ne doit pas être repris. Des tests de laboratoire pour déterminer les taux d'enzymes hépatiques doivent être effectués dès le premier symptôme ou signe de dysfonctionnement hépatique (p. Tests de laboratoire , INFORMATIONS PATIENTS ].
Événements cardiovasculaires graves
Mort subite et anomalies cardiaques structurelles préexistantes ou autres problèmes cardiaques graves
Enfants et adolescents
Des cas de mort subite ont été rapportés en association avec un traitement par atomoxétine aux doses habituelles chez des enfants et des adolescents présentant des anomalies cardiaques structurelles ou d'autres problèmes cardiaques graves. Bien que certains problèmes cardiaques graves comportent à eux seuls un risque accru de mort subite, l'atomoxétine ne doit généralement pas être utilisée chez les enfants ou les adolescents présentant des anomalies cardiaques structurelles graves connues, une cardiomyopathie, des anomalies graves du rythme cardiaque ou d'autres problèmes cardiaques graves qui peuvent les rendre plus vulnérables. aux effets noradrénergiques de l'atomoxétine.
Adultes
Des morts subites, des accidents vasculaires cérébraux et des infarctus du myocarde ont été signalés chez des adultes prenant de l'atomoxétine aux doses habituelles pour le TDAH. Bien que le rôle de l'atomoxétine dans ces cas adultes soit également inconnu, les adultes sont plus susceptibles que les enfants d'avoir des anomalies cardiaques structurelles graves, une cardiomyopathie, des anomalies graves du rythme cardiaque, une maladie coronarienne ou d'autres problèmes cardiaques graves. Il faut envisager de ne pas traiter les adultes présentant des anomalies cardiaques cliniquement significatives.
Évaluation de l'état cardiovasculaire chez les patients traités par l'atomoxétine
Les enfants, les adolescents ou les adultes pour lesquels un traitement par atomoxétine est envisagé doivent avoir une anamnèse minutieuse (y compris une évaluation des antécédents familiaux de mort subite ou d'arythmie ventriculaire) et un examen physique pour évaluer la présence d'une maladie cardiaque, et doivent recevoir d'autres examens cardiaques. évaluation si les résultats suggèrent une telle maladie (p. ex., électrocardiogramme et échocardiogramme). Les patients qui développent des symptômes tels que des douleurs thoraciques à l'effort, une syncope inexpliquée ou d'autres symptômes évoquant une maladie cardiaque pendant le traitement par l'atomoxétine doivent subir une évaluation cardiaque rapide.
Effets sur la tension artérielle et la fréquence cardiaque
STRATTERA doit être utilisé avec prudence chez les patients dont les conditions médicales sous-jacentes pourraient être aggravées par des augmentations de la pression artérielle ou de la fréquence cardiaque, tels que certains patients souffrant d'hypertension, de tachycardie ou de maladies cardiovasculaires ou cérébrovasculaires. Il ne doit pas être utilisé chez les patients présentant des troubles cardiaques ou vasculaires sévères dont l'état serait susceptible de se détériorer s'ils subissaient des augmentations cliniquement importantes de la pression artérielle ou de la fréquence cardiaque [voir CONTRE-INDICATIONS ]. Le pouls et la pression artérielle doivent être mesurés au départ, après les augmentations de dose de STRATTERA, et périodiquement pendant le traitement afin de détecter d'éventuelles augmentations cliniquement importantes.
Le tableau suivant fournit des données d'essais cliniques à court terme contrôlés par placebo pour les proportions de patients ayant une augmentation de : tension artérielle diastolique ≥ 15 mm Hg ; pression artérielle systolique ≥ 20 mm Hg ; fréquence cardiaque supérieure ou égale à 20 bpm, aussi bien dans la population pédiatrique que chez l'adulte (voir Tableau 1).
Dans les études d'enregistrement contrôlées par placebo portant sur des patients pédiatriques, la tachycardie a été identifiée comme un événement indésirable chez 0,3 % (5/1597) de ces patients recevant STRATTERA 10 mg, contre 0 % (0/934) des patients sous placebo. L'augmentation moyenne de la fréquence cardiaque chez les patients métaboliseurs rapides (EM) était de 5,0 battements/minute, et chez les patients métaboliseurs lents (PM) de 9,4 battements/minute.
Dans les essais cliniques adultes où le statut EM/PM était disponible, l'augmentation moyenne de la fréquence cardiaque chez les patients PM était significativement plus élevée que chez les patients EM (11 battements/minute contre 7,5 battements/minute). Les effets sur la fréquence cardiaque pourraient être cliniquement importants chez certains patients PM.
Dans les études d'enregistrement contrôlées par placebo impliquant des patients adultes, la tachycardie a été identifiée comme un événement indésirable chez 1,5 % (8/540) des patients recevant STRATTERA 25 mg, contre 0,5 % (2/402) des patients sous placebo.
Dans les essais cliniques adultes où le statut EM/PM était disponible, le changement moyen par rapport au départ de la pression artérielle diastolique chez les patients PM était plus élevé que chez les patients EM (4,21 contre 2,13 mm Hg), tout comme le changement moyen par rapport au départ de la pression artérielle systolique (PM : 2,75 contre EM : 2,40 mm Hg). Les effets sur la tension artérielle pourraient être cliniquement importants chez certains patients PM.
Une hypotension orthostatique et une syncope ont été signalées chez des patients prenant STRATTERA. Dans les études d'enregistrement d'enfants et d'adolescents, 0,2 % (12/5596) des patients traités par STRATTERA ont présenté une hypotension orthostatique et 0,8 % (46/5596) une syncope. Dans les études d'enregistrement à court terme d'enfants et d'adolescents, 1,8 % (6/340) des patients traités par STRATTERA ont présenté une hypotension orthostatique par rapport à 0,5 % (1/207) des patients sous placebo. Aucune syncope n'a été rapportée au cours des études d'enregistrement à court terme contrôlées par placebo sur le TDAH chez l'enfant et l'adolescent. STRATTERA 10 mg doit être utilisé avec prudence dans toute affection susceptible de prédisposer les patients à l'hypotension ou dans des affections associées à des changements brusques du rythme cardiaque ou de la tension artérielle.
Apparition de nouveaux symptômes psychotiques ou maniaques
Des symptômes psychotiques ou maniaques apparus sous traitement, par exemple des hallucinations, des idées délirantes ou une manie chez les enfants et les adolescents sans antécédents de maladie psychotique ou de manie, peuvent être provoqués par l'atomoxétine aux doses habituelles. Si de tels symptômes surviennent, il faut envisager un éventuel rôle causal de l'atomoxétine et l'arrêt du traitement doit être envisagé. Dans une analyse groupée de plusieurs études à court terme contrôlées par placebo, de tels symptômes sont survenus chez environ 0,2 % (4 patients présentant des réactions sur 1 939 exposés à l'atomoxétine pendant plusieurs semaines aux doses habituelles) des patients traités par l'atomoxétine, contre 0 sur 1 000. 1056 patients traités par placebo.
Dépistage des patients pour le trouble bipolaire
En général, des précautions particulières doivent être prises lors du traitement du TDAH chez les patients présentant un trouble bipolaire comorbide en raison du risque d'induction possible d'un épisode mixte/maniaque chez les patients à risque de trouble bipolaire. On ne sait pas si l'un des symptômes décrits ci-dessus représente une telle conversion. Cependant, avant d'initier le traitement par STRATTERA 40 mg, les patients présentant des symptômes dépressifs comorbides doivent faire l'objet d'un dépistage adéquat afin de déterminer s'ils présentent un risque de trouble bipolaire ; un tel dépistage devrait inclure des antécédents psychiatriques détaillés, y compris des antécédents familiaux de suicide, de trouble bipolaire et de dépression.
Comportement agressif ou hostilité
Les patients qui commencent un traitement pour le TDAH doivent être surveillés pour détecter l'apparition ou l'aggravation d'un comportement agressif ou d'hostilité. Un comportement agressif ou hostile est souvent observé chez les enfants et les adolescents atteints de TDAH. Dans les essais cliniques pédiatriques contrôlés à court terme, 21/1308 (1,6 %) des patients sous atomoxétine contre 9/806 (1,1 %) des patients sous placebo ont signalé spontanément des événements indésirables liés à l'hostilité liés au traitement (risque relatif global de 1,33 [95 % IC 0,67-2,64 – pas statistiquement significatif]). Dans des essais cliniques contrôlés par placebo chez des adultes, 6/1697 (0,35 %) des patients sous atomoxétine contre 4/1560 (0,26 %) des patients sous placebo ont signalé spontanément des événements indésirables liés à l'hostilité liés au traitement (risque relatif global de 1,38 [IC à 95 % 0,39-4,88 – pas statistiquement significatif]). Bien qu'il ne s'agisse pas d'une preuve concluante que STRATTERA 10 mg provoque un comportement agressif ou de l'hostilité, ces comportements ont été plus fréquemment observés dans les essais cliniques chez les enfants, les adolescents et les adultes traités par STRATTERA par rapport au placebo.
Événements allergiques
Bien que peu fréquentes, des réactions allergiques, y compris des réactions anaphylactiques, un œdème de Quincke, de l'urticaire et des éruptions cutanées, ont été signalées chez des patients prenant STRATTERA.
Effets sur la sortie d'urine de la vessie
Dans des essais contrôlés sur le TDAH chez l'adulte, les taux de rétention urinaire (1,7 %, 9/540) et d'hésitation urinaire (5,6 %, 30/540) ont augmenté chez les sujets sous atomoxétine par rapport aux sujets sous placebo (0 %, 0/402 ; 0,5 %, 2/402, respectivement). Deux sujets adultes sous atomoxétine et aucun sujet sous placebo ont abandonné les essais cliniques contrôlés en raison d'une rétention urinaire. Une plainte de rétention urinaire ou d'hésitation urinaire doit être considérée comme potentiellement liée à l'atomoxétine.
Priapisme
De rares cas post-commercialisation de priapisme, définis comme une érection pénienne douloureuse et non douloureuse durant plus de 4 heures, ont été rapportés chez des patients pédiatriques et adultes traités par STRATTERA. Les érections se sont résolues dans les cas où des informations de suivi étaient disponibles, certaines après l'arrêt de STRATTERA. Une attention médicale rapide est requise en cas de suspicion de priapisme.
Effets sur la croissance
Les données sur les effets à long terme de STRATTERA 25mg sur la croissance proviennent d'études en ouvert, et les variations de poids et de taille sont comparées aux données normatives de la population. En général, le gain de poids et de taille des patients pédiatriques traités par STRATTERA est inférieur à celui prédit par les données de population normatives pendant environ les 9 à 12 premiers mois de traitement. Par la suite, la prise de poids rebondit et à environ 3 ans de traitement, les patients traités par STRATTERA ont pris 17,9 kg en moyenne, soit 0,5 kg de plus que prévu par leurs données de base. Après environ 12 mois, la prise de taille se stabilise et à 3 ans, les patients traités par STRATTERA 40 mg ont gagné 19,4 cm en moyenne, soit 0,4 cm de moins que prévu par leurs données de base (voir Figure 1 ci-dessous).
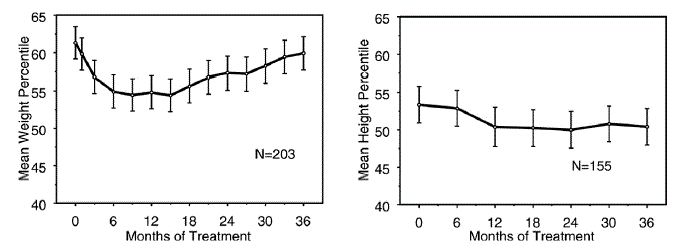
Figure 1 : Centiles moyens de poids et de taille au fil du temps pour les patients ayant reçu trois ans de traitement par STRATTERA à 40 mg
Ce modèle de croissance était généralement similaire quel que soit le statut pubertaire au moment de l'initiation du traitement. Les patients qui étaient prépubères au début du traitement (filles ≤ 8 ans, garçons ≤ 9 ans) ont pris en moyenne 2,1 kg et 1,2 cm de moins que prévu après trois ans. Les patients qui étaient pubères (filles > 8 à ≤ 13 ans, garçons > 9 à ≤ 14 ans) ou en fin de puberté (filles > 13 ans, garçons > 14 ans) avaient des gains de poids et de taille moyens proches ou ont dépassé ceux prévus après trois ans de traitement.
La croissance a suivi un schéma similaire chez les métaboliseurs rapides et lents (EM, PM). Les PM traités pendant au moins deux ans ont pris en moyenne 2,4 kg et 1,1 cm de moins que prévu, tandis que les EM ont pris en moyenne 0,2 kg et 0,4 cm de moins que prévu.
Dans des études contrôlées à court terme (jusqu'à 9 semaines), les patients traités par STRATTERA ont perdu en moyenne 0,4 kg et gagné en moyenne 0,9 cm, contre un gain de 1,5 kg et 1,1 cm chez les patients sous placebo. Dans un essai contrôlé à dose fixe, 1,3 %, 7,1 %, 19,3 % et 29,1 % des patients ont perdu au moins 3,5 % de leur poids corporel dans les groupes recevant le placebo, 0,5, 1,2 et 1,8 mg/kg/jour.
La croissance doit être surveillée pendant le traitement par STRATTERA.
Tests de laboratoire
Des tests de laboratoire de routine ne sont pas nécessaires.
Métabolisme du CYP2D6
Les métaboliseurs lents (MP) du CYP2D6 ont une ASC 10 fois supérieure et une concentration maximale 5 fois supérieure à une dose donnée de STRATTERA 25 mg par rapport aux métaboliseurs rapides (EM). Environ 7 % de la population caucasienne sont des PM. Des tests de laboratoire sont disponibles pour identifier les MP du CYP2D6. Les taux sanguins chez les PM sont similaires à ceux atteints en prenant de puissants inhibiteurs du CYP2D6. Les taux sanguins plus élevés chez les PM entraînent un taux plus élevé de certains effets indésirables de STRATTERA [voir EFFETS INDÉSIRABLES ].
Utilisation concomitante d'inhibiteurs puissants du CYP2D6 ou utilisation chez des patients connus pour être des PM du CYP2D6
L'atomoxétine est principalement métabolisée par la voie CYP2D6 en 4-hydroxyatomoxétine. Un ajustement posologique de STRATTERA peut être nécessaire lorsqu'il est co-administré avec de puissants inhibiteurs du CYP2D6 (par exemple, la paroxétine, la fluoxétine et la quinidine) ou lorsqu'il est administré à des MP du CYP2D6. [Voir DOSAGE ET ADMINISTRATION et INTERACTIONS MÉDICAMENTEUSES ].
Informations sur les conseils aux patients
Voir approuvé par la FDA INFORMATIONS PATIENTS .
Informations générales
Les médecins doivent demander à leurs patients de lire le Guide des médicaments avant de commencer le traitement par STRATTERA et de le relire chaque fois que la prescription est renouvelée.
Les prescripteurs ou autres professionnels de la santé doivent informer les patients, leurs familles et leurs soignants des bénéfices et des risques associés au traitement par STRATTERA et les conseiller sur son utilisation appropriée. Le prescripteur ou le professionnel de la santé doit demander aux patients, à leurs familles et à leurs soignants de lire le Guide des médicaments et doit les aider à comprendre son contenu. Les patients doivent avoir la possibilité de discuter du contenu du Guide de Médication et d'obtenir des réponses à toutes les questions qu'ils pourraient avoir.
Les patients doivent être informés des problèmes suivants et invités à alerter leur médecin prescripteur si ceux-ci surviennent pendant la prise de STRATTERA.
Risque suicidaire
Les patients, leurs familles et leurs soignants doivent être encouragés à être attentifs à l'émergence d'anxiété, d'agitation, d'attaques de panique, d'insomnie, d'irritabilité, d'hostilité, d'agressivité, d'impulsivité, d'akathisie (agitation psychomotrice), d'hypomanie, de manie, d'autres changements inhabituels de comportement , dépression et idées suicidaires, surtout au début du traitement par STRATTERA et lorsque la dose est ajustée. Les familles et les soignants des patients doivent être avisés d'observer l'apparition de tels symptômes au jour le jour, car les changements peuvent être brusques. De tels symptômes doivent être signalés au médecin prescripteur ou au professionnel de la santé du patient, en particulier s'ils sont graves, d'apparition brutale ou ne font pas partie des symptômes présentés par le patient. De tels symptômes peuvent être associés à un risque accru de pensées et de comportements suicidaires et indiquent la nécessité d'une surveillance très étroite et éventuellement de modifications de la médication.
Lésions hépatiques graves
Les patients qui commencent STRATTERA doivent être avertis qu'une atteinte hépatique grave peut se développer. Les patients doivent être informés qu'ils doivent contacter immédiatement leur médecin s'ils développent un prurit, une urine foncée, une jaunisse, une sensibilité de l'hypochondre droit ou des symptômes « pseudo-grippaux » inexpliqués [voir AVERTISSEMENTS ET PRECAUTIONS ].
Agression ou hostilité
Les patients doivent être informés qu'ils doivent appeler leur médecin dès que possible s'ils remarquent une augmentation de l'agressivité ou de l'hostilité.
Priapisme
De rares cas post-commercialisation de priapisme, définis comme une érection pénienne douloureuse et non douloureuse durant plus de 4 heures, ont été rapportés chez des patients pédiatriques et adultes traités par STRATTERA. Les parents ou les tuteurs des patients pédiatriques prenant STRATTERA 25 mg et des patients adultes prenant STRATTERA doivent être informés que le priapisme nécessite une attention médicale rapide.
Irritant oculaire
STRATTERA est un irritant oculaire. Les capsules STRATTERA ne sont pas destinées à être ouvertes. En cas de contact du contenu de la gélule avec l'œil, l'œil affecté doit être immédiatement rincé à l'eau et un avis médical doit être obtenu. Les mains et toutes les surfaces potentiellement contaminées doivent être lavées dès que possible.
Interaction médicament-médicament
Les patients doivent être informés qu'ils doivent consulter un médecin s'ils prennent ou prévoient de prendre des médicaments sur ordonnance ou en vente libre, des compléments alimentaires ou des remèdes à base de plantes.
Grossesse
Les patientes doivent être informées qu'elles doivent consulter un médecin si elles allaitent, sont enceintes ou envisagent de devenir enceintes pendant qu'elles prennent STRATTERA.
Aliments
Les patients peuvent prendre STRATTERA 25 mg avec ou sans nourriture.
Dose oubliée
Si les patients manquent une dose, ils doivent être informés de la prendre dès que possible, mais ne doivent pas prendre plus que la quantité quotidienne totale prescrite de STRATTERA sur une période de 24 heures.
Interférence avec les performances psychomotrices
Les patients doivent être informés de la prudence lorsqu'ils conduisent une voiture ou utilisent des machines dangereuses jusqu'à ce qu'ils soient raisonnablement certains que leurs performances ne sont pas affectées par l'atomoxétine.
Toxicologie non clinique
Carcinogenèse, mutagenèse, altération de la fertilité
Carcinogenèse
L'atomoxétine HCl n'était pas cancérigène chez les rats et les souris lorsqu'il était administré dans l'alimentation pendant 2 ans à des doses moyennes pondérées dans le temps allant jusqu'à 47 et 458 mg/kg/jour, respectivement. La dose la plus élevée utilisée chez le rat est d'environ 8 et 5 fois la dose humaine maximale chez l'enfant et l'adulte, respectivement, sur une base en mg/m2. Les taux plasmatiques (ASC) d'atomoxétine à cette dose chez le rat sont estimés à 1,8 fois (métaboliseurs rapides) ou 0,2 fois (métaboliseurs lents) ceux chez l'homme recevant la dose humaine maximale. La dose la plus élevée utilisée chez les souris est d'environ 39 et 26 fois la dose humaine maximale chez les enfants et les adultes, respectivement, sur une base mg/m2.
Mutagenèse
Le chlorhydrate d'atomoxétine s'est révélé négatif dans une batterie d'études de génotoxicité comprenant un test de mutation inverse (test d'Ames), un test in vitro de lymphome de souris, un test d'aberration chromosomique sur des cellules ovariennes de hamster chinois, un test de synthèse d'ADN non programmé sur des hépatocytes de rat et un test du micronoyau in vivo chez la souris. Cependant, il y avait une légère augmentation du pourcentage de cellules ovariennes de hamster chinois avec des diplochromosomes, suggérant une endoreduplication (aberration numérique).
Le métabolite N-desmethylatomoxetine HCl était négatif dans le test d'Ames, le test du lymphome de souris et le test de synthèse d'ADN non programmé.
Altération de la fertilité
L'atomoxétine HCl n'a pas altéré la fertilité chez le rat lorsqu'il a été administré dans l'alimentation à des doses allant jusqu'à 57 mg/kg/jour, soit environ 6 fois la dose maximale chez l'homme exprimée en mg/m2.
Utilisation dans des populations spécifiques
Grossesse
Catégorie de grossesse C
Des lapines gravides ont été traitées avec jusqu'à 100 mg/kg/jour d'atomoxétine par gavage tout au long de la période d'organogenèse. À cette dose, dans 1 des 3 études, une diminution du nombre de fœtus vivants et une augmentation des résorptions précoces ont été observées. De légères augmentations de l'incidence de l'origine atypique de l'artère carotide et de l'absence d'artère sous-clavière ont été observées. Ces résultats ont été observés à des doses qui provoquaient une légère toxicité maternelle. La dose sans effet pour ces résultats était de 30 mg/kg/jour. La dose de 100 mg/kg est environ 23 fois la dose humaine maximale sur une base mg/m2 ; les taux plasmatiques (ASC) d'atomoxétine à cette dose chez les lapins sont estimés à 3,3 fois (métaboliseurs rapides) ou 0,4 fois (métaboliseurs lents) ceux des humains recevant la dose humaine maximale.
Des rats ont été traités avec jusqu'à environ 50 mg/kg/jour d'atomoxétine (environ 6 fois la dose humaine maximale sur une base de mg/m2) dans l'alimentation à partir de 2 semaines (femelles) ou 10 semaines (mâles) avant l'accouplement jusqu'à la périodes d'organogenèse et de lactation. Dans 1 des 2 études, des diminutions du poids et de la survie des petits ont été observées. La diminution de la survie des ratons a également été observée à 25 mg/kg (mais pas à 13 mg/kg). Dans une étude au cours de laquelle des rats ont reçu de l'atomoxétine dans leur alimentation à partir de 2 semaines (femelles) ou de 10 semaines (mâles) avant l'accouplement tout au long de la période d'organogenèse, une diminution du poids fœtal (femelle uniquement) et une augmentation de l'incidence de une ossification incomplète de l'arc vertébral chez les fœtus a été observée à 40 mg/kg/jour (environ 5 fois la dose humaine maximale exprimée en mg/m2) mais pas à 20 mg/kg/jour.
Aucun effet fœtal indésirable n'a été observé lorsque des rats gravides ont été traités avec jusqu'à 150 mg/kg/jour (environ 17 fois la dose humaine maximale sur une base en mg/m2) par gavage tout au long de la période d'organogénèse. Aucune étude adéquate et bien contrôlée n'a été menée chez la femme enceinte. STRATTERA 18 mg ne doit pas être utilisé pendant la grossesse sauf si le bénéfice potentiel justifie le risque potentiel pour le fœtus.
Travail et accouchement
La parturition chez les rats n'a pas été affectée par l'atomoxétine. L'effet de STRATTERA sur le travail et l'accouchement chez l'homme est inconnu.
Mères allaitantes
L'atomoxétine et/ou ses métabolites ont été excrétés dans le lait de rats. On ne sait pas si l'atomoxétine est excrétée dans le lait maternel. La prudence s'impose si STRATTERA est administré à une femme qui allaite.
Utilisation pédiatrique
Toute personne envisageant l'utilisation de STRATTERA chez un enfant ou un adolescent doit équilibrer les risques potentiels avec le besoin clinique [voir AVERTISSEMENT DE BOÎTE et AVERTISSEMENTS ET PRECAUTIONS ]. La pharmacocinétique de l'atomoxétine chez les enfants et les adolescents est similaire à celle des adultes. L'innocuité, l'efficacité et la pharmacocinétique de STRATTERA chez les patients pédiatriques de moins de 6 ans n'ont pas été évaluées.
Une étude a été menée chez de jeunes rats pour évaluer les effets de l'atomoxétine sur la croissance et le développement neurocomportemental et sexuel. Des rats ont été traités avec 1, 10 ou 50 mg/kg/jour (environ 0,2, 2 et 8 fois, respectivement, la dose humaine maximale exprimée en mg/m2) d'atomoxétine administrée par gavage dès le début de la période postnatale (jour 10 ans) jusqu'à l'âge adulte. Légers retards d'apparition de la perméabilité vaginale (toutes doses) et de la séparation préputiale (10 et 50 mg/kg), légères diminutions du poids de l'épididyme et du nombre de spermatozoïdes (10 et 50 mg/kg), et légère diminution des corps jaunes (50 mg/kg /kg) ont été observés, mais il n'y a eu aucun effet sur la fertilité ou les performances de reproduction. Un léger retard dans le début de l'éruption des incisives a été observé à 50 mg/kg. Une légère augmentation de l'activité motrice a été observée au jour 15 (mâles à 10 et 50 mg/kg et femelles à 50 mg/kg) et au jour 30 (femelles à 50 mg/kg), mais pas au jour 60. Il n'y avait aucun effet sur les tests d'apprentissage et de mémoire. La signification de ces découvertes pour l'homme est inconnue.
Utilisation gériatrique
La sécurité, l'efficacité et la pharmacocinétique de STRATTERA 10 mg chez les patients gériatriques n'ont pas été évaluées.
Insuffisance hépatique
L'exposition à l'atomoxétine (AUC) est augmentée, par rapport aux sujets normaux, chez les sujets EM présentant une insuffisance hépatique modérée (Child-Pugh Classe B) (augmentation de 2 fois) et sévère (Child-Pugh Classe C) (augmentation de 4 fois). Un ajustement posologique est recommandé chez les patients présentant une insuffisance hépatique modérée ou sévère [voir DOSAGE ET ADMINISTRATION ].
Insuffisance rénale
Les sujets EM atteints d'insuffisance rénale terminale avaient une exposition systémique à l'atomoxétine plus élevée que les sujets sains (augmentation d'environ 65 %), mais il n'y avait pas de différence lorsque l'exposition était corrigée pour la dose en mg/kg. STRATTERA 25 mg peut donc être administré aux patients atteints de TDAH présentant une insuffisance rénale terminale ou des degrés moindres d'insuffisance rénale en utilisant le schéma posologique normal.
Le genre
Le sexe n'a pas influencé la disposition de l'atomoxétine.
Origine ethnique
L'origine ethnique n'a pas influencé la disposition de l'atomoxétine (sauf que les PM sont plus fréquentes chez les Caucasiens).
Patients atteints d'une maladie concomitante
Tics chez les patients atteints de TDAH et du trouble de la Tourette comorbide
L'atomoxétine administrée dans une gamme de doses flexibles de 0,5 à 1,5 mg/kg/jour (dose moyenne de 1,3 mg/kg/jour) et un placebo ont été comparés chez 148 sujets pédiatriques randomisés (âgés de 717 ans) avec un diagnostic DSM-IV de TDAH et tic comorbide dans une étude de 18 semaines, en double aveugle, contrôlée par placebo dans laquelle la majorité (80 %) s'est inscrite à cet essai avec le trouble de la Tourette (le trouble de la Tourette : 116 sujets ; le tic moteur chronique : 29 sujets). Une analyse de non-infériorité a révélé que STRATTERA 10 mg n'a pas aggravé les tics chez ces patients, tel que déterminé par le score total de l'échelle de sévérité des tics de Yale (YGTSS). Sur 148 patients entrés dans la phase aiguë du traitement, 103 (69,6 %) patients ont arrêté l'étude. La raison principale de l'arrêt dans les groupes de traitement par atomoxétine (38 patients sur 76, 50,0 %) et placebo (45 patients sur 72, 62,5 %) a été identifiée comme étant le manque d'efficacité, la plupart des patients ayant arrêté à la semaine 12. première visite où les patients avec un CGI-S≥4 pouvaient également répondre aux critères de « non-répondeur clinique » (le CGI-S est resté le même ou a augmenté par rapport au départ de l'étude) et être éligibles pour participer à une étude d'extension en ouvert avec l'atomoxétine. Il y a eu des rapports post-commercialisation de tics [voir EFFETS INDÉSIRABLES ].
Anxiété chez les patients atteints de TDAH et de troubles anxieux comorbides
Dans deux essais post-commercialisation, en double aveugle, contrôlés par placebo, il a été démontré que le traitement de patients atteints de TDAH et de troubles anxieux comorbides avec STRATTERA 10 mg n'aggrave pas leur anxiété.
Dans un essai contrôlé par placebo en double aveugle de 12 semaines, 176 patients, âgés de 8 à 17 ans, qui répondaient aux critères du DSM-IV pour le TDAH et au moins un des troubles anxieux du trouble d'anxiété de séparation, du trouble d'anxiété généralisée ou de la phobie sociale ont été aléatoire. Après une introduction en double aveugle contre placebo de 2 semaines, STRATTERA 25 mg a été initié à 0,8 mg/kg/jour avec augmentation à une dose cible de 1,2 mg/kg/jour (dose médiane 1,30 mg/kg/jour +/- 0,29 mg/kg/jour). STRATTERA n'a pas aggravé l'anxiété chez ces patients, tel que déterminé par l'échelle d'évaluation de l'anxiété pédiatrique (PARS). Sur les 158 patients qui ont terminé l'introduction en double aveugle avec placebo, 26 (16 %) patients ont arrêté l'étude.
Dans un essai séparé de 16 semaines, en double aveugle et contrôlé par placebo, 442 patients âgés de 18 à 65 ans, qui répondaient aux critères du DSM-IV pour le TDAH adulte et le trouble d'anxiété sociale (dont 23 % avaient également un trouble d'anxiété généralisée) ont été randomisés. Après une introduction de 2 semaines en double aveugle contre placebo, STRATTERA 10 mg a été initié à 40 mg/jour jusqu'à une dose maximale de 100 mg/jour (dose quotidienne moyenne 83 mg/jour +/- 19,5 mg/jour). STRATTERA 18 mg n'a pas aggravé l'anxiété chez ces patients, tel que déterminé par l'échelle d'anxiété sociale de Liebowitz (LSAS). Sur les 413 patients qui ont terminé l'introduction en double aveugle avec placebo, 149 (36,1 %) patients ont arrêté l'étude. Des cas d'anxiété ont été signalés après commercialisation [voir EFFETS INDÉSIRABLES ] .
SURDOSAGE
Expérience humaine
L'expérience des essais cliniques sur le surdosage de STRATTERA est limitée. Au cours de la post-commercialisation, des décès ont été signalés impliquant une surdose d'ingestion mixte de STRATTERA 10 mg et d'au moins un autre médicament. Il n'y a eu aucun rapport de décès impliquant un surdosage de STRATTERA seul, y compris des surdosages intentionnels à des doses allant jusqu'à 1400 mg. Dans certains cas de surdosage impliquant STRATTERA, des convulsions ont été rapportées. Les symptômes les plus fréquemment signalés accompagnant les surdosages aigus et chroniques de STRATTERA étaient les symptômes gastro-intestinaux, la somnolence, les étourdissements, les tremblements et les comportements anormaux. Une hyperactivité et une agitation ont également été rapportées. Des signes et symptômes compatibles avec une activation légère à modérée du système nerveux sympathique (p. ex., tachycardie, augmentation de la pression artérielle, mydriase, bouche sèche) ont également été observés. La plupart des événements étaient légers à modérés. Moins fréquemment, des cas d'allongement de l'intervalle QT et de changements mentaux, notamment une désorientation et des hallucinations, ont été rapportés [voir PHARMACOLOGIE CLINIQUE ].
Gestion du surdosage
Consultez un centre antipoison certifié pour obtenir des conseils et des conseils à jour. Étant donné que l'atomoxétine est fortement liée aux protéines, la dialyse n'est probablement pas utile dans le traitement d'un surdosage.
CONTRE-INDICATIONS
Hypersensibilité
STRATTERA est contre-indiqué chez les patients connus pour être hypersensibles à l'atomoxétine ou à d'autres constituants du produit [voir AVERTISSEMENTS ET PRECAUTIONS ].
Inhibiteurs de la monoamine oxydase (IMAO)
STRATTERA ne doit pas être pris avec un IMAO ou dans les 2 semaines suivant l'arrêt d'un IMAO. Le traitement par un IMAO ne doit pas être instauré dans les 2 semaines suivant l'arrêt de STRATTERA. Avec d'autres médicaments qui modifient les concentrations cérébrales de monoamines, des réactions graves, parfois mortelles, ont été rapportées (notamment hyperthermie, rigidité, myoclonie, instabilité autonome avec possibles fluctuations rapides des signes vitaux et modifications de l'état mental, notamment une agitation extrême évoluant vers le délire et coma) lorsqu'il est pris en association avec un IMAO. Certains cas présentaient des caractéristiques ressemblant au syndrome malin des neuroleptiques. De telles réactions peuvent survenir lorsque ces médicaments sont administrés simultanément ou à proximité [voir INTERACTIONS MÉDICAMENTEUSES ].
Glaucome à angle fermé
Dans les essais cliniques, l'utilisation de STRATTERA a été associée à un risque accru de mydriase et son utilisation n'est donc pas recommandée chez les patients atteints de glaucome à angle fermé.
Phéochromocytome
Des réactions graves, y compris une pression artérielle élevée et une tachyarythmie, ont été signalées chez des patients atteints de phéochromocytome ou ayant des antécédents de phéochromocytome qui ont reçu STRATTERA. Par conséquent, STRATTERA 18 mg ne doit pas être pris par des patients atteints de phéochromocytome ou ayant des antécédents de phéochromocytome.
Troubles cardiovasculaires graves
STRATTERA 40 mg ne doit pas être utilisé chez les patients présentant des troubles cardiaques ou vasculaires sévères dont l'état est susceptible de se détériorer s'ils présentent des augmentations de la pression artérielle ou de la fréquence cardiaque pouvant être cliniquement importantes (par exemple, 15 à 20 mm Hg de tension artérielle ou 20 battements par minute de fréquence cardiaque). [Voir AVERTISSEMENTS ET PRECAUTIONS ].
PHARMACOLOGIE CLINIQUE
Mécanisme d'action
Le mécanisme précis par lequel l'atomoxétine produit ses effets thérapeutiques dans le trouble déficitaire de l'attention/hyperactivité (TDAH) est inconnu, mais on pense qu'il est lié à l'inhibition sélective du transporteur présynaptique de la noradrénaline, comme déterminé dans des études ex vivo sur l'absorption et l'épuisement des neurotransmetteurs .
Pharmacodynamie
Une analyse exposition-réponse englobant des doses d'atomoxétine (0,5, 1,2 ou 1,8 mg/kg/jour) ou un placebo a démontré que l'exposition à l'atomoxétine est corrélée à l'efficacité, telle que mesurée par l'échelle d'évaluation du trouble de déficit de l'attention/hyperactivité-IV-version parentale : l'investigateur a administré et marqué. La relation exposition-efficacité était similaire à celle observée entre la dose et l'efficacité, les expositions médianes aux deux doses les plus élevées entraînant des changements presque maximaux par rapport à la valeur initiale [voir Etudes cliniques ].
Électrophysiologie cardiaque
L'effet de STRATTERA sur l'allongement de l'intervalle QTc a été évalué dans une étude croisée, randomisée, en double aveugle, positive (moxifloxacine 400 mg) et contrôlée par placebo chez des hommes sains métaboliseurs lents du CYP2D6. Au total, 120 sujets sains ont reçu STRATTERA (20 mg et 60 mg) deux fois par jour pendant 7 jours. Aucune modification importante de l'intervalle QTc (c.-à-d. augmentations > 60 msec par rapport à la ligne de base, QTc absolu > 480 msec) n'a été observée dans l'étude. Cependant, de petits changements dans l'intervalle QTc ne peuvent pas être exclus de l'étude actuelle, car l'étude n'a pas réussi à démontrer la sensibilité du test. Il y avait une légère augmentation de l'intervalle QTc avec l'augmentation de la concentration d'atomoxétine.
Pharmacocinétique
L'atomoxétine est bien absorbée après administration orale et est peu affectée par les aliments. Il est éliminé principalement par métabolisme oxydatif via la voie enzymatique du cytochrome P450 2D6 (CYP2D6) et par glucuronidation subséquente. L'atomoxétine a une demi-vie d'environ 5 heures. Une fraction de la population (environ 7 % des Caucasiens et 2 % des Afro-Américains) sont des métaboliseurs lents (MP) des médicaments métabolisés par le CYP2D6. Ces personnes ont une activité réduite dans cette voie, ce qui entraîne des ASC 10 fois plus élevées, des concentrations plasmatiques maximales 5 fois plus élevées et une élimination plus lente (demi-vie plasmatique d'environ 24 heures) de l'atomoxétine par rapport aux personnes ayant une activité normale [métaboliseurs rapides (EM )]. Les médicaments qui inhibent le CYP2D6, tels que la fluoxétine, la paroxétine et la quinidine, provoquent des augmentations similaires de l'exposition.
La pharmacocinétique de l'atomoxétine a été évaluée chez plus de 400 enfants et adolescents dans des essais cliniques sélectionnés, principalement à l'aide d'études pharmacocinétiques de population. Des données pharmacocinétiques individuelles à dose unique et à l'état d'équilibre ont également été obtenues chez les enfants, les adolescents et les adultes. Lorsque les doses ont été normalisées en mg/kg, des valeurs similaires de demi-vie, de Cmax et d'ASC ont été observées chez les enfants, les adolescents et les adultes. La clairance et le volume de distribution après ajustement pour le poids corporel étaient également similaires.
Absorption et distribution
L'atomoxétine est rapidement absorbée après administration orale, avec une biodisponibilité absolue d'environ 63 % chez les EM et de 94 % chez les PM. Les concentrations plasmatiques maximales (Cmax) sont atteintes environ 1 à 2 heures après l'administration.
STRATTERA 18 mg peut être administré avec ou sans nourriture. L'administration de STRATTERA avec un repas standard riche en graisses chez l'adulte n'a pas affecté le degré d'absorption orale de l'atomoxétine (AUC), mais a diminué le taux d'absorption, entraînant une Cmax inférieure de 37 % et un Tmax retardé de 3 heures. Dans les essais cliniques menés chez des enfants et des adolescents, l'administration de STRATTERA avec de la nourriture a entraîné une diminution de 9 % de la Cmax.
Le volume de distribution à l'état d'équilibre après administration intraveineuse est de 0,85 L/kg, ce qui indique que l'atomoxétine se distribue principalement dans l'eau corporelle totale. Le volume de distribution est similaire dans toute la plage de poids du patient après normalisation pour le poids corporel.
Aux concentrations thérapeutiques, 98 % de l'atomoxétine dans le plasma est liée aux protéines, principalement l'albumine.
Métabolisme et élimination
L'atomoxétine est principalement métabolisée par la voie enzymatique CYP2D6. Les personnes ayant une activité réduite dans cette voie (PM) ont des concentrations plasmatiques d'atomoxétine plus élevées que les personnes ayant une activité normale (EM). Pour les PM, l'ASC de l'atomoxétine est d'environ 10 fois et la Css, max est d'environ 5 fois supérieure à celle des EM. Des tests de laboratoire sont disponibles pour identifier les MP du CYP2D6. L'administration concomitante de STRATTERA avec des inhibiteurs puissants du CYP2D6, tels que la fluoxétine, la paroxétine ou la quinidine, entraîne une augmentation substantielle de l'exposition plasmatique à l'atomoxétine, et un ajustement posologique peut être nécessaire [voir AVERTISSEMENTS ET PRECAUTIONS ]. L'atomoxétine n'a pas inhibé ni induit la voie CYP2D6.
Le principal métabolite oxydatif formé, quel que soit le statut CYP2D6, est la 4-hydroxyatomoxétine, qui est glucuronidée. La 4-hydroxyatomoxétine est équipotente à l'atomoxétine en tant qu'inhibiteur du transporteur de noradrénaline mais circule dans le plasma à des concentrations beaucoup plus faibles (1 % de la concentration d'atomoxétine dans les EM et 0,1 % de la concentration d'atomoxétine dans les PM). La 4-hydroxyatomoxétine est principalement formée par le CYP2D6, mais dans les PM, la 4-hydroxyatomoxétine est formée à un rythme plus lent par plusieurs autres enzymes du cytochrome P450. La N-desméthylatomoxétine est formée par le CYP2C19 et d'autres enzymes du cytochrome P450, mais a une activité pharmacologique nettement inférieure à celle de l'atomoxétine et circule dans le plasma à des concentrations plus faibles (5 % de la concentration d'atomoxétine dans les EM et 45 % de la concentration d'atomoxétine dans les PM).
La clairance plasmatique apparente moyenne de l'atomoxétine après administration orale chez l'adulte EM est de 0,35 L/h/kg et la demi-vie moyenne est de 5,2 heures. Suite à l'administration orale d'atomoxétine à des PM, la clairance plasmatique apparente moyenne est de 0,03 L/h/kg et la demi-vie moyenne est de 21,6 heures. Pour les PM, l'ASC de l'atomoxétine est d'environ 10 fois et la Css, max est d'environ 5 fois supérieure à celle des EM. La demi-vie d'élimination de la 4-hydroxyatomoxétine est similaire à celle de la N-desméthylatomoxétine (6 à 8 heures) chez les sujets EM, tandis que la demi-vie de la N-desméthylatomoxétine est beaucoup plus longue chez les sujets PM (34 à 40 heures).
L'atomoxétine est principalement excrétée sous forme de 4-hydroxyatomoxétine-O-glucuronide, principalement dans l'urine (plus de 80 % de la dose) et, dans une moindre mesure, dans les fèces (moins de 17 % de la dose). Seule une petite fraction de la dose de STRATTERA 40 mg est excrétée sous forme d'atomoxétine inchangée (moins de 3 % de la dose), ce qui indique une biotransformation importante.
[Voir Utilisation dans des populations spécifiques ].
Etudes cliniques
Études sur le TDAH chez les enfants et les adolescents
Études aiguës
L'efficacité de STRATTERA 40 mg dans le traitement du TDAH a été établie dans 4 études randomisées, en double aveugle, contrôlées par placebo chez des patients pédiatriques (âgés de 6 à 18 ans). Environ un tiers des patients répondaient aux critères du DSM-IV pour le sous-type inattentif et les deux tiers répondaient aux critères des sous-types inattentifs et hyperactifs/impulsifs.
Les signes et les symptômes du TDAH ont été évalués par une comparaison du changement moyen entre le départ et le critère d'évaluation pour les patients traités par STRATTERA et par placebo à l'aide d'une analyse en intention de traiter du critère de jugement principal, l'investigateur a administré et noté l'échelle d'évaluation du TDAH-IV- Score total de la version parentale (ADHDRS) comprenant les sous-échelles hyperactif/impulsif et inattentif. Chaque élément de l'ADHDRS correspond directement à un critère de symptôme du TDAH dans le DSM-IV.
Dans l'étude 1, une étude de 8 semaines randomisée, en double aveugle, contrôlée par placebo, dose-réponse, sur le traitement aigu des enfants et des adolescents âgés de 8 à 18 ans (N = 297), les patients ont reçu soit une dose fixe de STRATTERA (0,5, 1,2 ou 1,8 mg/kg/jour) ou un placebo. STRATTERA 10 mg a été administré en dose fractionnée tôt le matin et en fin d'après-midi/début de soirée. Aux 2 doses plus élevées, les améliorations des symptômes du TDAH étaient statistiquement significativement supérieures chez les patients traités par STRATTERA par rapport aux patients traités par placebo, telles que mesurées sur l'échelle ADHDRS. La dose de 1,8 mg/kg/jour de STRATTERA 25 mg n'a pas apporté de bénéfice supplémentaire par rapport à celui observé avec la dose de 1,2 mg/kg/jour. La dose de 0,5 mg/kg/jour de STRATTERA n'a pas été supérieure au placebo.
Dans l'étude 2, une étude de traitement aigu randomisée, en double aveugle, contrôlée par placebo, d'une durée de 6 semaines menée auprès d'enfants et d'adolescents âgés de 6 à 16 ans (N = 171), les patients ont reçu soit STRATTERA 40 mg, soit un placebo. STRATTERA a été administré en une dose unique tôt le matin et titré en fonction du poids en fonction de la réponse clinique, jusqu'à une dose maximale de 1,5 mg/kg/jour. La dose finale moyenne de STRATTERA 10 mg était d'environ 1,3 mg/kg/jour. Les symptômes du TDAH ont été améliorés de manière statistiquement significative avec STRATTERA par rapport au placebo, tel que mesuré sur l'échelle ADHDRS. Cette étude montre que STRATTERA est efficace lorsqu'il est administré une fois par jour le matin.
Dans 2 études identiques, de 9 semaines, aiguës, randomisées, en double aveugle, contrôlées par placebo chez des enfants âgés de 7 à 13 ans (étude 3, N = 147 ; étude 4, N = 144), STRATTERA 40 mg et le méthylphénidate ont été comparés à un placebo . STRATTERA 18 mg a été administré en dose fractionnée tôt le matin et en fin d'après-midi (après l'école) et titré sur une base ajustée au poids en fonction de la réponse clinique. La dose maximale recommandée de STRATTERA était de 2,0 mg/kg/jour. La dose finale moyenne de STRATTERA 18 mg pour les deux études était d'environ 1,6 mg/kg/jour. Dans les deux études, les symptômes du TDAH se sont statistiquement significativement améliorés davantage avec STRATTERA 10 mg qu'avec le placebo, tel que mesuré sur l'échelle ADHDRS.
L'examen des sous-ensembles de population en fonction du sexe et de l'âge (
Étude d'entretien
L'efficacité de STRATTERA 18 mg dans le traitement d'entretien du TDAH a été établie dans une étude ambulatoire chez des enfants et des adolescents (âgés de 6 à 15 ans). Les patients répondant aux critères du DSM-IV pour le TDAH qui ont montré une réponse continue pendant environ 4 semaines au cours d'une phase initiale de traitement en ouvert de 10 semaines avec STRATTERA (1,2 à 1,8 mg/kg/jour) ont été randomisés pour continuer à prendre leur dose actuelle de STRATTERA (N =292) ou à un placebo (N=124) sous traitement en double aveugle pour l'observation d'une rechute. La réponse au cours de la phase en ouvert a été définie comme un score CGI-ADHD-S ≤ 2 et une réduction d'au moins 25 % par rapport au départ du score total ADHDRS-IV-Parent:Inv. Les patients qui ont été assignés à STRATTERA et ont montré une réponse continue pendant environ 8 mois au cours de la première phase de traitement en double aveugle ont été à nouveau randomisés pour continuer à prendre leur dose actuelle de STRATTERA (N = 81) ou pour recevoir un placebo (N = 82) en double aveugle. traitement pour l'observation de la rechute. La rechute au cours de la phase en double aveugle a été définie comme une augmentation du score CGI-TDAH-S d'au moins 2 à partir de la fin de la phase en ouvert et le score total ADHDRS-IV-Parent:Inv revient à ≥ 90 % du score d'entrée dans l'étude pour 2 visites consécutives. Dans les deux phases en double aveugle, les patients recevant un traitement continu par STRATTERA ont connu des temps de rechute significativement plus longs que ceux recevant le placebo.
Études sur le TDAH chez les adultes
L'efficacité de STRATTERA dans le traitement du TDAH a été établie dans 2 études cliniques randomisées, en double aveugle, contrôlées par placebo chez des patients adultes, âgés de 18 ans et plus, qui répondaient aux critères du DSM-IV pour le TDAH.
Les signes et les symptômes du TDAH ont été évalués à l'aide de la version de dépistage de l'échelle d'évaluation du TDAH pour adultes de Conners (CAARS), une échelle de 30 items administrée par l'investigateur. La principale mesure d'efficacité était le score total des symptômes du TDAH en 18 points (la somme des sous-échelles d'inattention et d'hyperactivité/impulsivité du CAARS) évalué par une comparaison du changement moyen entre le début et le point final à l'aide d'une analyse en intention de traiter.
Dans 2 études de traitement aigu identiques, randomisées, en double aveugle, contrôlées par placebo, d'une durée de 10 semaines (étude 5, N = 280 ; étude 6, N = 256), les patients ont reçu soit STRATTERA 25 mg, soit un placebo. STRATTERA a été administré en dose fractionnée tôt le matin et en fin d'après-midi/début de soirée et titré en fonction de la réponse clinique dans une fourchette de 60 à 120 mg/jour. La dose finale moyenne de STRATTERA 40 mg pour les deux études était d'environ 95 mg/jour. Dans les deux études, les symptômes du TDAH ont été améliorés de manière statistiquement significative avec STRATTERA, tel que mesuré sur le score des symptômes du TDAH de l'échelle CAARS.
L'examen des sous-ensembles de population en fonction du sexe et de l'âge (
INFORMATIONS PATIENTS
STRATTERA® (Stra-TAIR-a) (atomoxétine) Capsules
Lisez le Guide des médicaments qui accompagne STRATTERA® avant que vous ou votre enfant ne commenciez à le prendre et chaque fois que vous recevez une recharge. Il peut y avoir de nouvelles informations. Ce Guide de Médication ne prend pas l'endroit de parler à votre docteur de votre traitement ou du traitement de votre enfant avec STRATTERA.
Quelle est l'information la plus importante que je devrais connaître sur STRATTERA 18mg ?
Les effets suivants ont été rapportés avec l'utilisation de STRATTERA :
Les enfants et les adolescents pensent parfois au suicide et nombre d'entre eux déclarent avoir tenté de se suicider. Les résultats des études cliniques STRATTERA avec plus de 2200 enfants ou adolescents atteints de TDAH suggèrent que certains enfants et adolescents peuvent avoir un risque plus élevé d'avoir des pensées ou des actions suicidaires. Bien qu'aucun suicide ne soit survenu dans ces études, 4 patients sur 1000 ont développé des pensées suicidaires. Informez le médecin de votre enfant ou adolescent si votre enfant ou adolescent (ou s'il y a des antécédents familiaux de) :
- une maladie bipolaire (maladie maniaco-dépressive)
- avait des pensées ou des actions suicidaires avant de commencer STRATTERA
Le risque de pensées et d'actions suicidaires peut être plus élevé :
- tôt pendant le traitement par STRATTERA 10mg
- lors des ajustements posologiques
Prévenez les pensées et actions suicidaires chez votre enfant ou adolescent en :
- porter une attention particulière aux humeurs, comportements, pensées et sentiments de votre enfant ou adolescent pendant le traitement par STRATTERA 25 mg
- garder toutes les visites de suivi avec le médecin de votre enfant ou adolescent comme prévu
Surveillez les signes suivants chez votre enfant ou adolescent pendant le traitement par STRATTERA 25 mg :
- anxiété
- agitation
- crises de panique
- troubles du sommeil
- irritabilité
- hostilité
- agressivité
- impulsivité
- agitation
- la manie
- la dépression
- pensées suicidaires
Appelez immédiatement le médecin de votre enfant ou adolescent s'il présente l'un des signes ci-dessus, en particulier s'il est nouveau, soudain ou grave. Votre enfant ou adolescent peut avoir besoin d'être surveillé de près pour des pensées et des actions suicidaires ou avoir besoin d'un changement de médicament.
STRATTERA peut causer des lésions hépatiques chez certains patients. Appelez immédiatement votre médecin si vous ou votre enfant présentez les signes suivants de problèmes hépatiques :
- démangeaison
- douleur dans le haut du ventre droit
- urine foncée
- peau ou yeux jaunes
- symptômes pseudo-grippaux inexpliqués
- mort subite chez les patients qui ont des problèmes cardiaques ou des malformations cardiaques
- accident vasculaire cérébral et crise cardiaque chez les adultes
- augmentation de la pression artérielle et de la fréquence cardiaque
Informez votre médecin si vous ou votre enfant avez des problèmes cardiaques, des malformations cardiaques, de l'hypertension artérielle ou des antécédents familiaux de ces problèmes. Votre médecin doit vérifier attentivement si vous ou votre enfant avez des problèmes cardiaques avant de commencer STRATTERA.
Votre médecin doit vérifier régulièrement votre tension artérielle ou la tension artérielle et la fréquence cardiaque de votre enfant pendant le traitement par STRATTERA.
Appelez immédiatement votre médecin si vous ou votre enfant présentez des signes de problèmes cardiaques tels que douleurs thoraciques, essoufflement ou évanouissement pendant la prise de STRATTERA.
- de nouveaux symptômes psychotiques (comme entendre des voix, croire des choses qui ne sont pas vraies, être suspect) ou de nouveaux symptômes maniaques
Appelez immédiatement le médecin de votre enfant ou de votre adolescent au sujet de tout nouveau symptôme mental car un ajustement ou un arrêt du traitement par STRATTERA 40 mg peut être envisagé.
Qu'est-ce que STRATTERA 18mg?
STRATTERA est un médicament inhibiteur sélectif du recaptage de la noradrénaline. Il est utilisé pour le traitement du trouble déficitaire de l'attention et de l'hyperactivité (TDAH). STRATTERA peut aider à augmenter l'attention et à diminuer l'impulsivité et l'hyperactivité chez les patients atteints de TDAH.
STRATTERA 10 mg doit être utilisé dans le cadre d'un programme de traitement total du TDAH pouvant inclure des conseils ou d'autres thérapies.
STRATTERA n'a pas été étudié chez les enfants de moins de 6 ans.
Qui ne devrait pas prendre STRATTERA ?
STRATTERA ne doit pas être pris si vous ou votre enfant :
- prenez ou avez pris au cours des 14 derniers jours un médicament anti-dépression appelé inhibiteur de la monoamine oxydase ou IMAO. Certains noms de médicaments IMAO sont Nardil® (sulfate de phénelzine), Parnate® (sulfate de tranylcypromine) et Emsam® (système transdermique de sélégiline).
- avez un problème oculaire appelé glaucome à angle fermé
- sont allergiques à quoi que ce soit dans STRATTERA. Voir la fin de ce Guide de Médication pour une liste complète d'ingrédients.
- avez ou avez eu une tumeur rare appelée phéochromocytome.
STRATTERA peut ne pas convenir à vous ou à votre enfant. Avant de commencer STRATTERA, informez votre médecin ou le médecin de votre enfant de tous les problèmes de santé (ou d'antécédents familiaux), y compris :
- avoir ou avoir eu des pensées ou des actions suicidaires
- problèmes cardiaques, malformations cardiaques, rythme cardiaque irrégulier, hypertension artérielle ou hypotension artérielle
- problèmes mentaux, psychose, manie, maladie bipolaire ou dépression
- problèmes de foie Informez votre médecin si vous ou votre enfant êtes enceinte, planifiez une grossesse ou allaitez.
STRATTERA peut-il être pris avec d'autres médicaments ?
Informez votre médecin de tous les médicaments que vous ou votre enfant prenez, y compris les médicaments sur ordonnance et en vente libre, les vitamines et les suppléments à base de plantes. STRATTERA et certains médicaments peuvent interagir entre eux et provoquer des effets indésirables graves. Votre médecin décidera si STRATTERA peut être pris avec d'autres médicaments.
Informez en particulier votre médecin si vous ou votre enfant prenez :
- médicaments contre l'asthme
- les médicaments anti-dépression, y compris les IMAO
- médicaments contre l'hypertension
- médicaments contre le rhume ou les allergies contenant des décongestionnants
Connaissez les médicaments que vous ou votre enfant prenez. Gardez une liste de vos médicaments avec vous pour la montrer à votre médecin et à votre pharmacien.
Ne commencez pas de nouveau médicament pendant que vous prenez STRATTERA sans en parler d'abord à votre médecin.
Comment prendre STRATTERA ?
- Prenez STRATTERA 40 mg exactement comme prescrit. STRATTERA 18mg est disponible en gélules de différentes doses. Votre médecin peut ajuster la dose jusqu'à ce qu'elle soit appropriée pour vous ou votre enfant.
- Ne pas mâcher, écraser ou ouvrir les gélules. Avalez les capsules STRATTERA entières avec de l'eau ou d'autres liquides. Informez votre médecin si vous ou votre enfant ne pouvez pas avaler STRATTERA 10 mg en entier. Un autre médicament devra peut-être être prescrit.
- Évitez de toucher une gélule cassée de STRATTERA 18 mg. Se laver les mains et les surfaces qui ont touché une gélule ouverte de STRATTERA 10 mg. Si de la poudre entre en contact avec vos yeux ou ceux de votre enfant, rincez-les immédiatement à l'eau et appelez votre médecin.
- STRATTERA peut être pris avec ou sans nourriture.
- STRATTERA 10 mg est généralement pris une ou deux fois par jour. Prenez STRATTERA 25 mg à la même heure chaque jour pour vous aider à vous souvenir. Si vous manquez une dose de STRATTERA, prenez-la dès que vous vous souvenez de ce jour. Si vous manquez une journée de STRATTERA 10 mg, ne doublez pas votre dose le lendemain. Sautez simplement la journée que vous avez manquée.
- De temps en temps, votre médecin peut arrêter le traitement par STRATTERA 25 mg pendant un certain temps pour vérifier les symptômes du TDAH.
- Votre médecin peut effectuer des contrôles réguliers du sang, du cœur et de la tension artérielle pendant la prise de STRATTERA. Les enfants doivent faire vérifier souvent leur taille et leur poids pendant qu'ils prennent STRATTERA. Le traitement par STRATTERA pourra être arrêté si un problème est constaté lors de ces bilans.
- Si vous ou votre enfant prenez trop de STRATTERA ou faites une surdose, appelez immédiatement votre médecin ou un centre antipoison, ou obtenez un traitement d'urgence.
Quels sont les effets secondaires possibles de STRATTERA ?
Voir "Quelle est l'information la plus importante que je devrais connaître sur STRATTERA 10mg ?" pour obtenir des informations sur les pensées et actions suicidaires signalées, les autres problèmes mentaux, les lésions hépatiques graves et les problèmes cardiaques.
D'autres effets secondaires graves comprennent:
- réactions allergiques graves (appelez votre médecin si vous avez du mal à respirer, si vous voyez un gonflement ou de l'urticaire ou si vous présentez d'autres réactions allergiques)
- ralentissement de la croissance (taille et poids) chez les enfants
- problèmes pour uriner, y compris
- difficulté à démarrer ou à maintenir un jet d'urine
- ne peut pas vider complètement la vessie
Les effets secondaires courants chez les enfants et les adolescents comprennent :
- maux d'estomac
- diminution de l'appétit
- nausées ou vomissements
- vertiges
- fatigue
- sautes d'humeur
Les effets secondaires courants chez les adultes comprennent :
- constipation
- bouche sèche
- nausée
- diminution de l'appétit
- vertiges
- effets secondaires sexuels
- problèmes pour uriner
Autres informations pour les enfants, les adolescents et les adultes :
- Des érections persistantes (priapisme) sont survenues rarement pendant le traitement par STRATTERA. Si vous avez une érection qui dure plus de 4 heures, consultez immédiatement un médecin. En raison du potentiel de dommages durables, y compris l'incapacité potentielle d'avoir des érections, le priapisme doit être évalué par un médecin immédiatement.
- STRATTERA 10 mg peut affecter votre capacité ou celle de votre enfant à conduire ou à utiliser des machines lourdes. Soyez prudent jusqu'à ce que vous sachiez comment STRATTERA 25 mg vous affecte ou affecte votre enfant.
- Parlez à votre médecin si vous ou votre enfant avez des effets secondaires gênants ou qui ne disparaissent pas.
Ce n'est pas une liste complète des effets secondaires possibles. Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
Comment dois-je conserver STRATTERA ?
- Conservez STRATTERA dans un endroit sûr à température ambiante, entre 59 et 86 °F (15 et 30 °C).
- Gardez STRATTERA 40 mg et tous les médicaments hors de la portée des enfants.
Informations générales sur STRATTERA
Les médicaments sont parfois prescrits à des fins autres que celles énumérées dans un guide des médicaments. N'utilisez pas STRATTERA 25 mg pour une affection pour laquelle il n'a pas été prescrit. Ne donnez pas STRATTERA à d'autres personnes, même si elles ont la même condition. Cela peut leur nuire.
Ce Guide de Médication résume les renseignements les plus importants sur STRATTERA. Si vous souhaitez plus d'informations, parlez-en à votre médecin. Vous pouvez demander à votre médecin ou à votre pharmacien des informations sur STRATTERA qui ont été rédigées pour les professionnels de la santé. Pour plus d'informations sur STRATTERA, appelez le 1-800-Lilly-Rx (1-800-545-5979) ou visitez www.strattera.com.
Quels sont les ingrédients de STRATTERA 40mg ?
Ingrédient actif: chlorhydrate d'atomoxétine.
Ingrédients inactifs: amidon prégélatinisé, diméthicone, gélatine, laurylsulfate de sodium, FD&CBlue n° 2, oxyde de fer jaune synthétique, dioxyde de titane, oxyde de fer rouge et encre noire comestible.
Ce guide de médication a été approuvé par la Food and Drug Administration des États-Unis.