Protonix 20mg, 40mg Pantoprazole Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce que Protonix 40 mg et comment est-il utilisé ?
Protonix 40 mg est un médicament sur ordonnance utilisé pour traiter les symptômes de l'œsophagite causée par l'acide gastrique due au reflux gastro-œsophagien ou au RGO et au syndrome de Zollinger-Ellison ou à d'autres affections provoquant un excès d'acide gastrique. Protonix 40 mg peut être utilisé seul ou avec d'autres médicaments.
Protonix appartient à une classe de médicaments appelés inhibiteurs de la pompe à protons (IPP).
On ne sait pas si Protonix est sûr et efficace chez les enfants de moins de 5 ans.
Quels sont les effets secondaires possibles de Protonix ?
Protonix peut provoquer des effets secondaires graves, notamment :
- fortes douleurs à l'estomac,
- diarrhée aqueuse ou contenant du sang,
- douleur soudaine ou difficulté à bouger la hanche, le poignet ou le dos,
- ecchymose ou gonflement au site d'injection,
- peu ou pas de miction,
- sang dans les urines,
- gonflement,
- prise de poids rapide,
- vertiges,
- rythme cardiaque rapide ou irrégulier,
- tremblements (tremblements) ou mouvements saccadés,
- se sentir nerveux,
- crampes ou spasmes musculaires dans les mains ou les pieds,
- toux,
- sensation d'étouffement,
- douleurs articulaires et
- éruption cutanée sur les joues ou les bras qui s'aggrave la nuit
Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires les plus courants de Protonix incluent :
- mal de tête,
- vertiges,
- Douleur d'estomac,
- gaz,
- nausée,
- vomissement,
- diarrhée,
- douleur articulaire,
- fièvre,
- éruption cutanée, et
- symptômes du rhume (plus fréquents chez les enfants)
Dites à votre médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles de Prometrium. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
LA DESCRIPTION
L'ingrédient actif de PROTONIX (pantoprazole sodique) pour suspension orale à libération retardée et de PROTONIX (pantoprazole sodique) comprimés à libération retardée, un IPP, est un benzimidazole substitué, sodium 5-(difluorométhoxy)-2-[[(3,4- diméthoxy-2- pyridinyl)méthyl] sulfinyl]-1H-benzimidazole sesquihydraté, un composé qui inhibe la sécrétion d'acide gastrique. Sa formule empirique est C16H14F2N3NaO4S x 1,5 H2O, avec un poids moléculaire de 432,4. La formule structurale est :
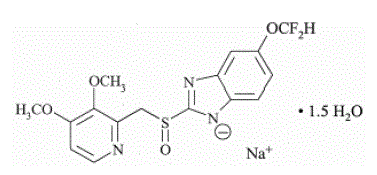
Le pantoprazole sodique sesquihydraté est une poudre cristalline blanche à blanc cassé et est racémique. Le pantoprazole a des propriétés faiblement basiques et acides. Le pantoprazole sodique sesquihydraté est librement soluble dans l'eau, très légèrement soluble dans un tampon phosphate à pH 7,4 et pratiquement insoluble dans le n-hexane.
La stabilité du composé en solution aqueuse dépend du pH. Le taux de dégradation augmente avec la diminution du pH. A température ambiante, la demi-vie de dégradation est d'environ 2,8 heures à pH 5 et d'environ 220 heures à pH 7,8.
PROTONIX 40 mg est fourni sous forme de suspension buvable à libération retardée en sachets unitaires, disponible en un seul dosage (40 mg) de pantoprazole (équivalent à 45,1 mg de pantoprazole sodique), et sous forme de comprimé à libération retardée, disponible en deux dosages 20 mg de pantoprazole (équivalent à 22,56 mg de pantoprazole sodique) et 40 mg de pantoprazole (équivalent à 45,1 mg de pantoprazole sodique).
Les comprimés à libération retardée PROTONIX contiennent les ingrédients inactifs suivants : stéarate de calcium, crospovidone, hypromellose, oxyde de fer, mannitol, copolymère d'acide méthacrylique, polysorbate 80, povidone, propylène glycol, carbonate de sodium, laurylsulfate de sodium, dioxyde de titane et citrate de triéthyle. Les comprimés à libération retardée PROTONIX (40 mg et 20 mg) sont conformes au test de dissolution USP 2.
PROTONIX 40 mg pour suspension orale à libération retardée contient les ingrédients inactifs suivants : crospovidone, hypromellose, copolymère d'acide méthacrylique, cellulose microcristalline, polysorbate 80, povidone, carbonate de sodium, laurylsulfate de sodium, talc, dioxyde de titane, citrate de triéthyle et oxyde de fer jaune.
LES INDICATIONS
PROTONIX pour suspension orale à libération retardée et PROTONIX 40 mg comprimés à libération retardée sont indiqués pour :
Traitement à court terme de l'œsophagite érosive associée au reflux gastro-œsophagien (RGO)
PROTONIX 20 mg est indiqué chez les adultes et les patients pédiatriques de cinq ans et plus pour le traitement à court terme (jusqu'à 8 semaines) dans la guérison et le soulagement symptomatique de l'œsophagite érosive (EE). Pour les patients adultes qui n'ont pas guéri après 8 semaines de traitement, une cure supplémentaire de 8 semaines de PROTONIX peut être envisagée. La sécurité du traitement au-delà de 8 semaines chez les patients pédiatriques n'a pas été établie.
Entretien de la guérison de l'oesophagite érosive
PROTONIX est indiqué pour le maintien de la guérison de l'EE et la réduction des taux de rechute des symptômes de brûlures d'estomac diurnes et nocturnes chez les patients adultes atteints de RGO. Les études contrôlées ne se sont pas prolongées au-delà de 12 mois.
Conditions hypersécrétoires pathologiques, y compris le syndrome de Zollinger-Ellison
PROTONIX 20 mg est indiqué pour le traitement à long terme des conditions hypersécrétoires pathologiques, y compris le syndrome de Zollinger-Ellison (ZE).
DOSAGE ET ADMINISTRATION
Programme de dosage recommandé
PROTONIX est fourni sous forme de granulés à libération retardée en sachets pour la préparation de suspensions buvables ou sous forme de comprimés à libération retardée. Les doses recommandées sont décrites dans le tableau 1.
Consignes administratives
Le mode d'administration pour chaque forme posologique est présenté dans le tableau 2.
Prenez une dose oubliée dès que possible. S'il est presque l'heure de la dose suivante, sautez la dose oubliée et prenez la dose suivante à l'heure prévue. Ne prenez pas 2 doses en même temps.
Comprimés à libération retardée PROTONIX
Avalez les comprimés à libération retardée PROTONIX entiers, avec ou sans nourriture dans l'estomac. Pour les patients incapables d'avaler un comprimé de 40 mg, deux comprimés de 20 mg peuvent être pris. L'administration concomitante d'antiacides n'affecte pas l'absorption des comprimés à libération retardée PROTONIX.
PROTONIX pour suspension orale à libération retardée
Administrez PROTONIX pour suspension orale à libération retardée environ 30 minutes avant un repas par voie orale dans du jus de pomme ou de la compote de pommes ou par sonde nasogastrique dans du jus de pomme uniquement. Étant donné qu'un pH approprié est nécessaire pour la stabilité, ne pas administrer PROTONIX 20 mg pour suspension orale à libération retardée dans des liquides autres que le jus de pomme ou des aliments autres que la compote de pommes.
Ne pas diviser le paquet de 40 mg de PROTONIX 20 mg pour suspension orale à libération retardée pour créer une dose de 20 mg pour les patients pédiatriques qui ne peuvent pas prendre la formulation en comprimés.
PROTONIX pour suspension orale à libération retardée - administration orale dans la compote de pommes
- Paquet ouvert.
- Saupoudrer de granulés sur une cuillerée à café de compote de pommes. NE PAS UTILISER D'AUTRES ALIMENTS OU ÉCRASER OU MÂCHER LES GRANULES.
- Prendre dans les 10 minutes suivant la préparation.
- Prenez des gorgées d'eau pour vous assurer que les granulés sont lavés dans l'estomac. Répétez les gorgées d'eau si nécessaire.
PROTONIX pour suspension orale à libération retardée - administration orale dans du jus de pomme
- Paquet ouvert.
- Videz les granulés dans une petite tasse ou une cuillère à café contenant une cuillère à café de jus de pomme.
- Remuer pendant 5 secondes (les granules ne se dissolvent pas) et avaler immédiatement.
- Pour vous assurer que la dose entière est prise, rincez le récipient une ou deux fois avec du jus de pomme pour éliminer les granules restants. Avalez immédiatement.
PROTONIX 40 mg pour suspension orale à libération retardée - Administration d'une sonde nasogastrique (NG) ou d'une sonde de gastrostomie
Pour les patients qui ont une sonde nasogastrique ou une sonde de gastrostomie en place, PROTONIX 40 mg pour suspension orale à libération retardée peut être administré comme suit :
- Retirez le piston du corps d'une seringue à pointe de cathéter de 2 onces (60 ml). Jeter le piston.
- Connectez l'extrémité du cathéter de la seringue à un tube de 16 French (ou plus).
- Tenez la seringue attachée à la tubulure aussi haut que possible tout en administrant PROTONIX pour suspension orale à libération retardée afin d'éviter toute flexion de la tubulure.
- Videz le contenu du sachet dans le corps de la seringue.
- Ajouter 10 mL (2 cuillerées à thé) de jus de pomme et tapoter doucement et/ou secouer le cylindre de la seringue pour aider à rincer la seringue et le tube. Répétez au moins deux fois de plus en utilisant la même quantité de jus de pomme (10 ml ou 2 cuillerées à thé) à chaque fois. Aucun granulé ne doit rester dans la seringue.
COMMENT FOURNIE
Formes posologiques et points forts
Comprimés à libération retardée :
- 40 mg de pantoprazole, comprimés jaunes ovales biconvexes portant l'inscription PROTONIX (encre brune) sur une face
- 20 mg de pantoprazole, comprimés jaunes ovales biconvexes portant l'inscription P20 (encre brune) sur une face
Pour la suspension orale à libération retardée :
- 40 mg de pantoprazole, granulés à enrobage entérique de couleur jaunâtre pâle à brun foncé dans un sachet à dose unitaire
Stockage et manutention
PROTONIX (pantoprazole sodique) Comprimés à libération retardée sont fournis sous forme de comprimés à libération retardée jaunes, ovales et biconvexes portant l'inscription PROTONIX (encre brune) sur une face contenant 40 mg de pantoprazole et sont disponibles comme suit :
CDN 0008-0841-81, bouteilles de 90
PROTONIX (pantoprazole sodique) Comprimés à libération retardée sont fournis sous forme de comprimés à libération retardée biconvexes ovales jaunes portant l'inscription P20 (encre brune) sur une face contenant 20 mg de pantoprazole et sont disponibles comme suit :
CDN 0008-0843-81, bouteilles de 90
PROTONIX (pantoprazole sodique) pour suspension orale à libération retardée est fourni sous forme de granulés à enrobage entérique de couleur jaunâtre pâle à brun foncé contenant 40 mg de pantoprazole dans un sachet-dose et est disponible comme suit :
CDN 0008-0844-02, carton unidose de 30
Stockage
Conservez PROTONIX 40 mg pour suspension orale à libération retardée et PROTONIX 20 mg comprimés à libération retardée entre 20 et 25 °C (68 et 77 °F); excursions permises jusqu'à 15° à 30°C (59° à 86°F) [voir Température ambiante contrôlée USP ].
Distribué par : Wyeth Pharmaceuticals LLC, une filiale de Pfizer Inc., Philadelphie, PA 19101. Révisé : novembre 2020
EFFETS SECONDAIRES
Les effets indésirables graves suivants sont décrits ci-dessous et ailleurs dans l'étiquetage :
- Néphrite tubulo-interstitielle aiguë [voir AVERTISSEMENTS ET PRECAUTIONS ]
- Diarrhée associée à Clostridium difficile [voir AVERTISSEMENTS ET PRECAUTIONS ]
- Fracture osseuse [voir AVERTISSEMENTS ET PRECAUTIONS ]
- Lupus érythémateux cutané et disséminé [voir AVERTISSEMENTS ET PRECAUTIONS ]
- Carence en cyanocobalamine (vitamine B-12) [voir AVERTISSEMENTS ET PRECAUTIONS ]
- Hypomagnésémie [voir AVERTISSEMENTS ET PRECAUTIONS ]
- Polypes de la glande fundique [voir AVERTISSEMENTS ET PRECAUTIONS ]
Expérience des essais cliniques
Les profils d'effets indésirables de PROTONIX (pantoprazole sodique) pour suspension orale à libération retardée et de PROTONIX (pantoprazole sodique) comprimés à libération retardée sont similaires.
Étant donné que les essais cliniques sont menés dans des conditions très variables, les taux d'effets indésirables observés dans les essais cliniques d'un médicament ne peuvent pas être directement comparés aux taux des essais cliniques d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique clinique.
Adultes
L'innocuité dans neuf essais cliniques comparatifs randomisés aux États-Unis chez des patients atteints de RGO comprenait 1 473 patients sous PROTONIX oral (20 mg ou 40 mg), 299 patients sous un antagoniste des récepteurs H2, 46 patients sous un autre IPP et 82 patients sous placebo. Les effets indésirables les plus fréquents sont répertoriés dans le tableau 3.
Les effets indésirables supplémentaires qui ont été signalés pour PROTONIX 20 mg dans les essais cliniques avec une fréquence ≤ 2 % sont énumérés ci-dessous par système corporel :
Corps dans son ensemble : réaction allergique, pyrexie, réaction de photosensibilité, œdème facial
Gastro-intestinal : constipation, bouche sèche, hépatite
Hématologique : leucopénie, thrombocytopénie
Métabolique/Nutritionnel : CK élevée (créatine kinase), œdème généralisé, triglycérides élevés, enzymes hépatiques élevées
Musculo-squelettique : myalgie
Nerveux: dépression, vertige
Peau et appendices : urticaire, rash, prurit
Sens spéciaux : Vision floue
Patients pédiatriques
L'innocuité de PROTONIX dans le traitement de l'EE associée au RGO a été évaluée chez des patients pédiatriques âgés de 1 an à 16 ans dans trois essais cliniques. Les essais d'innocuité impliquaient des patients pédiatriques atteints d'EE ; cependant, comme l'EE est rare dans la population pédiatrique, 249 patients pédiatriques atteints de RGO symptomatique ou prouvé par endoscopie ont également été évalués. Tous les effets indésirables de PROTONIX 20 mg chez l'adulte sont considérés comme pertinents pour les patients pédiatriques. Chez les patients âgés de 1 an à 16 ans, les effets indésirables les plus fréquemment rapportés (> 4 %) incluent : URI, maux de tête, fièvre, diarrhée, vomissements, éruption cutanée et douleurs abdominales.
Pour les informations de sécurité chez les patients de moins de 1 an, voir Utilisation dans des populations spécifiques .
Les effets indésirables supplémentaires qui ont été signalés pour PROTONIX 20 mg chez les patients pédiatriques dans les essais cliniques avec une fréquence ≤ 4 % sont énumérés ci-dessous par système corporel :
Corps dans son ensemble : réaction allergique, œdème facial
Gastro-intestinal : constipation, flatulences, nausées
Métabolique/Nutritionnel : taux élevé de triglycérides, taux élevé d'enzymes hépatiques, taux élevé de CK (créatine kinase)
Musculo-squelettique : arthralgie, myalgie
Nerveux: étourdissement, vertige
Peau et appendices : urticaire
Les effets indésirables suivants observés chez les adultes dans les essais cliniques n'ont pas été rapportés chez les patients pédiatriques dans les essais cliniques, mais sont considérés comme pertinents pour les patients pédiatriques : réaction de photosensibilité, bouche sèche, hépatite, thrombocytopénie, œdème généralisé, dépression, prurit, leucopénie et vision trouble. .
Syndrome de Zollinger-Ellison (ZE)
Dans les études cliniques sur le syndrome ZE, les effets indésirables rapportés chez 35 patients prenant PROTONIX 80 mg/jour à 240 mg/jour pendant 2 ans au maximum étaient similaires à ceux rapportés chez les patients adultes atteints de RGO.
Expérience post-commercialisation
Les effets indésirables suivants ont été identifiés lors de l'utilisation post-approbation de PROTONIX. Étant donné que ces réactions sont signalées volontairement par une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale avec l'exposition au médicament.
Ces effets indésirables sont répertoriés ci-dessous par système corporel :
Problèmes gastro-intestinaux: polypes des glandes fundiques
Troubles généraux et conditions d'administration : asthénie, fatigue, malaise
Hématologique : pancytopénie, agranulocytose
Troubles hépatobiliaires : lésions hépatocellulaires entraînant un ictère et une insuffisance hépatique
Troubles du système immunitaire : anaphylaxie (y compris choc anaphylactique), lupus érythémateux disséminé
Infections et infestations : Diarrhée associée à Clostridium difficile
Enquêtes : changements de poids
Troubles du métabolisme et de la nutrition : hyponatrémie, hypomagnésémie
Troubles musculo-squelettiques: rhabdomyolyse, fracture osseuse
Nerveux: agueusie, dysgueusie
Troubles psychiatriques: hallucination, confusion, insomnie, somnolence
Troubles rénaux et urinaires : néphrite tubulo-interstitielle aiguë
Troubles de la peau et du tissu sous-cutané : réactions dermatologiques sévères (certaines mortelles), y compris érythème polymorphe, syndrome de Stevens-Johnson, nécrolyse épidermique toxique (TEN, certaines mortelles), angio-œdème (œdème de Quincke) et lupus érythémateux cutané
INTERACTIONS MÉDICAMENTEUSES
Le tableau 4 comprend les médicaments ayant des interactions médicamenteuses cliniquement importantes et des interactions avec les diagnostics lorsqu'ils sont administrés en concomitance avec PROTONIX et des instructions pour les prévenir ou les gérer.
Consultez l'étiquetage des médicaments utilisés en concomitance pour obtenir de plus amples informations sur les interactions avec les IPP.
AVERTISSEMENTS
Inclus dans le cadre du PRÉCAUTIONS section.
PRÉCAUTIONS
Présence de malignité gastrique
Chez l'adulte, la réponse symptomatique au traitement par PROTONIX n'exclut pas la présence d'une tumeur maligne gastrique. Envisager un suivi et des tests diagnostiques supplémentaires chez les patients adultes qui ont une réponse sous-optimale ou une rechute symptomatique précoce après avoir terminé le traitement avec un IPP. Chez les patients plus âgés, envisagez également une endoscopie.
Néphrite tubulo-interstitielle aiguë
Une néphrite tubulo-interstitielle (TIN) aiguë a été observée chez des patients prenant des IPP et peut survenir à tout moment au cours du traitement par IPP. Les patients peuvent présenter divers signes et symptômes allant de réactions d'hypersensibilité symptomatiques à des symptômes non spécifiques d'une diminution de la fonction rénale (p. ex., malaise, nausées, anorexie). Dans les séries de cas rapportés, certains patients ont été diagnostiqués sur biopsie et en l'absence de manifestations extra-rénales (p. ex., fièvre, éruption cutanée ou arthralgie). Arrêtez PROTONIX 20 mg et évaluez les patients chez qui on soupçonne une TIN aiguë [voir CONTRE-INDICATIONS ].
Diarrhée associée à Clostridium difficile
Des études observationnelles publiées suggèrent qu'un traitement par IPP tel que PROTONIX 20 mg peut être associé à un risque accru de diarrhée associée à Clostridium difficile, en particulier chez les patients hospitalisés. Ce diagnostic doit être évoqué en cas de diarrhée qui ne s'améliore pas [voir EFFETS INDÉSIRABLES ].
Les patients doivent utiliser la dose la plus faible et la durée la plus courte de traitement par IPP appropriées à l'affection traitée.
Fracture de l'os
Plusieurs études observationnelles publiées suggèrent que le traitement par IPP peut être associé à un risque accru de fractures de la hanche, du poignet ou de la colonne vertébrale liées à l'ostéoporose. Le risque de fracture était accru chez les patients ayant reçu une dose élevée, définie comme de multiples doses quotidiennes, et un traitement par IPP à long terme (un an ou plus). Les patients doivent utiliser la dose la plus faible et la durée la plus courte de traitement par IPP appropriées à l'affection traitée. Les patients à risque de fractures liées à l'ostéoporose doivent être pris en charge conformément aux directives de traitement établies [voir DOSAGE ET ADMINISTRATION , EFFETS INDÉSIRABLES ].
Lupus érythémateux cutané et disséminé
Des cas de lupus érythémateux cutané (CLE) et de lupus érythémateux disséminé (LES) ont été signalés chez des patients prenant des IPP, y compris le pantoprazole sodique. Ces événements se sont produits à la fois comme une nouvelle apparition et comme une exacerbation d'une maladie auto-immune existante. La majorité des cas de lupus érythémateux induits par les IPP étaient des CLE.
La forme la plus courante de CLE signalée chez les patients traités par des IPP était la CLE subaiguë (SCLE) et s'est produite dans les semaines ou les années suivant un traitement médicamenteux continu chez des patients allant des nourrissons aux personnes âgées. Généralement, les résultats histologiques ont été observés sans atteinte des organes.
Le lupus érythémateux disséminé (LES) est moins fréquemment rapporté que le CLE chez les patients recevant des IPP. Le LED associé aux IPP est généralement plus léger que le LED non médicamenteux. L'apparition du LES s'est généralement produite quelques jours à quelques années après le début du traitement, principalement chez des patients allant des jeunes adultes aux personnes âgées. La majorité des patients présentaient une éruption cutanée ; cependant, des arthralgies et des cytopénies ont également été signalées.
Évitez l'administration d'IPP plus longtemps que médicalement indiqué. Si des signes ou des symptômes compatibles avec le CLE ou le SLE sont notés chez les patients recevant PROTONIX 40 mg, arrêtez le médicament et référez le patient au spécialiste approprié pour évaluation. La plupart des patients s'améliorent avec l'arrêt de l'IPP seul en 4 à 12 semaines. Les tests sérologiques (par exemple ANA) peuvent être positifs et les résultats des tests sérologiques élevés peuvent prendre plus de temps à se résoudre que les manifestations cliniques.
Carence en cyanocobalamine (vitamine B-12)
Généralement, un traitement quotidien avec des médicaments antiacides sur une longue période (par exemple, plus de 3 ans) peut entraîner une malabsorption de la cyanocobalamine (vitamine B-12) causée par une hypo ou une achlorhydrie. De rares cas de carence en cyanocobalamine survenant avec un traitement anti-acide ont été rapportés dans la littérature. Ce diagnostic doit être envisagé si des symptômes cliniques compatibles avec une carence en cyanocobalamine sont observés.
Hypomagnésémie
De rares cas d'hypomagnésémie, symptomatique et asymptomatique, ont été rapportés chez des patients traités par des IPP pendant au moins trois mois, et dans la plupart des cas après un an de traitement. Les événements indésirables graves comprennent la tétanie, les arythmies et les convulsions. Chez la plupart des patients, le traitement de l'hypomagnésémie a nécessité un remplacement du magnésium et l'arrêt de l'IPP.
Pour les patients qui devraient suivre un traitement prolongé ou qui prennent des IPP avec des médicaments tels que la digoxine ou des médicaments susceptibles de provoquer une hypomagnésémie (p. EFFETS INDÉSIRABLES ].
Tumorigénicité
En raison de la nature chronique du RGO, il peut y avoir un risque d'administration prolongée de PROTONIX. Dans des études à long terme sur des rongeurs, le pantoprazole était cancérigène et provoquait des types rares de tumeurs gastro-intestinales. La pertinence de ces résultats pour le développement de tumeurs chez l'homme est inconnue [voir Toxicologie non clinique ].
Polypes de la glande fundique
L'utilisation d'IPP est associée à un risque accru de polypes des glandes fundiques qui augmente avec une utilisation à long terme, en particulier au-delà d'un an. La plupart des utilisateurs d'IPP qui ont développé des polypes de la glande fundique étaient asymptomatiques et les polypes de la glande fundique ont été identifiés accidentellement à l'endoscopie. Utilisez la durée la plus courte de traitement par IPP appropriée à la condition traitée.
Interférence avec les enquêtes sur les tumeurs neuroendocrines
Les taux sériques de chromogranine A (CgA) augmentent en raison de la diminution de l'acidité gastrique induite par les médicaments. L'augmentation du taux de CgA peut entraîner des résultats faussement positifs dans les investigations diagnostiques des tumeurs neuroendocrines. Les professionnels de la santé doivent temporairement arrêter le traitement par PROTONIX 20 mg au moins 14 jours avant d'évaluer les taux de CgA et envisager de répéter le test si les taux initiaux de CgA sont élevés. Si des tests en série sont effectués (par exemple pour la surveillance), le même laboratoire commercial doit être utilisé pour les tests, car les plages de référence entre les tests peuvent varier [voir PHARMACOLOGIE CLINIQUE ].
Interférence avec l'écran d'urine pour le THC
Il y a eu des rapports de tests de dépistage urinaires faussement positifs pour le tétrahydrocannabinol (THC) chez des patients recevant des IPP, y compris PROTONIX [voir INTERACTIONS MÉDICAMENTEUSES ].
Utilisation concomitante de PROTONIX avec le méthotrexate
La littérature suggère que l'utilisation concomitante d'IPP avec le méthotrexate (principalement à forte dose ; voir les informations de prescription du méthotrexate) peut élever et prolonger les taux sériques de méthotrexate et/ou de son métabolite, entraînant éventuellement des toxicités du méthotrexate. En cas d'administration de méthotrexate à forte dose, un arrêt temporaire de l'IPP peut être envisagé chez certains patients [voir INTERACTIONS MÉDICAMENTEUSES ].
Informations sur les conseils aux patients
Conseillez au patient de lire l'étiquetage patient approuvé par la FDA ( Guide des médicaments et mode d'emploi ).
Malignité gastrique
Conseillez aux patients de retourner chez leur fournisseur de soins de santé s'ils ont une réponse sous-optimale ou une rechute symptomatique précoce [voir AVERTISSEMENTS ET PRECAUTIONS ].
Néphrite tubulo-interstitielle aiguë
Conseillez aux patients d'appeler immédiatement leur fournisseur de soins de santé s'ils présentent des signes et/ou des symptômes associés à une néphrite tubulo-interstitielle aiguë [voir CONTRE-INDICATIONS , AVERTISSEMENTS ET PRECAUTIONS ].
Diarrhée associée à Clostridium difficile
Conseillez aux patients d'appeler immédiatement leur fournisseur de soins de santé s'ils souffrent de diarrhée qui ne s'améliore pas [voir AVERTISSEMENTS ET PRECAUTIONS ].
Fracture de l'os
Conseillez aux patients de signaler toute fracture, en particulier de la hanche, du poignet ou de la colonne vertébrale, à leur fournisseur de soins de santé [voir AVERTISSEMENTS ET PRECAUTIONS ].
Lupus érythémateux cutané et disséminé
Conseillez aux patients d'appeler immédiatement leur fournisseur de soins de santé en cas d'apparition ou d'aggravation des symptômes associés au lupus érythémateux cutané ou disséminé [voir AVERTISSEMENTS ET PRECAUTIONS ].
Carence en cyanocobalamine (vitamine B-12)
Conseillez aux patients de signaler tout symptôme clinique pouvant être associé à une carence en cyancobalamine à leur professionnel de la santé s'ils reçoivent PROTONIX 40 mg depuis plus de 3 ans [voir AVERTISSEMENTS ET PRECAUTIONS ].
Hypomagnésémie
Conseillez aux patients de signaler tout symptôme clinique pouvant être associé à une hypomagnésémie à leur professionnel de la santé, s'ils reçoivent PROTONIX 40 mg depuis au moins 3 mois [voir AVERTISSEMENTS ET PRECAUTIONS ].
Interactions médicamenteuses
Demandez aux patients d'informer leur fournisseur de soins de santé de tout autre médicament qu'ils prennent actuellement, y compris les produits contenant de la rilpivirine [voir CONTRE-INDICATIONS ] digoxine [voir AVERTISSEMENTS ET PRECAUTIONS ] et méthotrexate à haute dose [voir AVERTISSEMENTS ET PRECAUTIONS ].
Grossesse
Informez une femme enceinte du risque potentiel pour le fœtus. Conseillez aux femmes en âge de procréer d'informer leur fournisseur de soins de santé d'une grossesse connue ou suspectée [voir Utilisation dans des populations spécifiques ].
Administration
- Ne pas diviser, écraser ou mâcher PROTONIX pour suspension orale à libération retardée et PROTONIX 20 mg comprimés à libération retardée.
- Le sachet de suspension buvable PROTONIX est une dose fixe et ne peut pas être divisé pour obtenir une dose plus petite.
- Avalez PROTONIX 20 mg comprimés à libération retardée entier, avec ou sans nourriture dans l'estomac.
- L'administration concomitante d'antiacides n'affecte pas l'absorption des comprimés à libération retardée PROTONIX.
- Prenez PROTONIX 20 mg pour suspension orale à libération retardée environ 30 minutes avant un repas.
- Administrez PROTONIX pour suspension orale à libération retardée dans du jus de pomme ou de la compote de pommes, comme décrit dans le mode d'emploi. Ne pas administrer dans l'eau, d'autres liquides ou aliments.
- Pour les patients porteurs d'une sonde nasogastrique (NG) ou de gastrostomie, PROTONIX pour suspension orale à libération retardée peut être administré avec du jus de pomme, comme décrit dans le mode d'emploi.
- Prenez une dose oubliée dès que possible. S'il est presque l'heure de la dose suivante, sautez la dose oubliée et prenez la dose suivante à l'heure prévue. Ne prenez pas 2 doses en même temps.
Toxicologie non clinique
Carcinogenèse, mutagenèse, altération de la fertilité
Dans une étude de cancérogénicité de 24 mois, des rats Sprague-Dawley ont été traités par voie orale avec des doses de pantoprazole de 0,5 à 200 mg/kg/jour, soit environ 0,1 à 40 fois l'exposition sur la surface corporelle d'une personne de 50 kg recevant 40 mg /journée. Dans le fundus gastrique, un traitement avec 0,5 à 200 mg/kg/jour a produit une hyperplasie des cellules de type entérochromaffine (ECL) et des tumeurs bénignes et malignes des cellules neuroendocrines d'une manière liée à la dose. Dans le préestomac, un traitement avec 50 et 200 mg/kg/jour (environ 10 et 40 fois la dose humaine recommandée sur la base de la surface corporelle) a produit des papillomes épidermoïdes bénins et des carcinomes épidermoïdes malins. Les tumeurs gastro-intestinales rares associées au traitement par le pantoprazole comprenaient un adénocarcinome du duodénum avec 50 mg/kg/jour et des polypes bénins et des adénocarcinomes du fundus gastrique avec 200 mg/kg/jour. Dans le foie, le traitement avec 0,5 à 200 mg/kg/jour a produit des augmentations liées à la dose de l'incidence des adénomes et des carcinomes hépatocellulaires. Dans la glande thyroïde, le traitement avec 200 mg/kg/jour a entraîné une incidence accrue d'adénomes et de carcinomes des cellules folliculaires chez les rats mâles et femelles.
Dans une étude de cancérogénicité de 24 mois, des rats Fischer 344 ont été traités par voie orale avec des doses de 5 à 50 mg/kg/jour de pantoprazole, soit environ 1 à 10 fois la dose humaine recommandée en fonction de la surface corporelle. Dans le fundus gastrique, un traitement avec 5 à 50 mg/kg/jour a produit une hyperplasie des cellules de type entérochromaffine (ECL) et des tumeurs bénignes et malignes des cellules neuroendocrines. La sélection de dose pour cette étude n'a peut-être pas été adéquate pour évaluer de manière exhaustive le potentiel carcinogène du pantoprazole.
Dans une étude de cancérogénicité de 24 mois, des souris B6C3F1 ont été traitées par voie orale avec des doses de 5 à 150 mg/kg/jour de pantoprazole, 0,5 à 15 fois la dose humaine recommandée en fonction de la surface corporelle. Dans le foie, le traitement avec 150 mg/kg/jour a entraîné une incidence accrue d'adénomes et de carcinomes hépatocellulaires chez les souris femelles. Le traitement avec 5 à 150 mg/kg/jour a également produit une hyperplasie des cellules ECL gastrique-fundique.
Une étude de cancérogénicité de 26 semaines sur des souris transgéniques p53 +/- n'a pas été positive.
Le pantoprazole s'est révélé positif dans les tests in vitro d'aberration chromosomique des lymphocytes humains, dans l'un des deux tests du micronoyau chez la souris pour les effets clastogènes et dans le test de mutation directe in vitro sur les cellules ovariennes de hamster chinois/HGPRT pour les effets mutagènes. Des résultats équivoques ont été observés dans le test de liaison covalente de l'ADN de foie de rat in vivo. Le pantoprazole s'est révélé négatif dans le test de mutation d'Ames in vitro, le test de synthèse d'ADN non programmée (UDS) in vitro avec des hépatocytes de rat, le test de mutation génique directe AS52/GPT de mammifère in vitro, le test de mutation de la thymidine kinase in vitro avec le lymphome de souris L5178Y cellules, et le test in vivo d'aberration chromosomique des cellules de moelle osseuse de rat.
Il n'y a eu aucun effet sur la fertilité ou les performances de reproduction lorsque le pantoprazole a été administré à des doses orales allant jusqu'à 500 mg/kg/jour chez les rats mâles (98 fois la dose humaine recommandée en fonction de la surface corporelle) et 450 mg/kg/jour chez les rats femelles. (88 fois la dose humaine recommandée basée sur la surface corporelle).
Utilisation dans des populations spécifiques
Grossesse
Résumé des risques
Les données disponibles provenant d'études observationnelles publiées n'ont pas démontré d'association de malformations majeures ou d'autres issues défavorables de la grossesse avec le pantoprazole.
Dans les études de reproduction chez l'animal, aucune preuve d'effets indésirables sur le développement n'a été observée avec le pantoprazole. Des études de reproduction ont été réalisées chez des rats à des doses orales allant jusqu'à 450 mg/kg/jour (environ 88 fois la dose humaine recommandée) et des lapins à des doses orales allant jusqu'à 40 mg/kg/jour (environ 16 fois la dose humaine recommandée) avec l'administration de pantoprazole au cours de l'organogenèse chez les animaux gravides et n'ont révélé aucun signe d'effet nocif sur le fœtus dû au pantoprazole dans cette étude (voir Données ).
Une étude de toxicité sur le développement prénatal et postnatal chez le rat avec des critères d'évaluation supplémentaires pour évaluer l'effet sur le développement osseux a été réalisée avec le pantoprazole sodique. Des doses orales de pantoprazole de 5, 15 et 30 mg/kg/jour (environ 1, 3 et 6 fois la dose humaine de 40 mg/jour) ont été administrées à des femmes enceintes du 6e jour de gestation (JG) au jour de lactation (LD ) 21. Des modifications de la morphologie osseuse ont été observées chez les ratons exposés au pantoprazole in utero et par le lait pendant la période de lactation ainsi que par voie orale du jour postnatal (PND) 4 au PND 21 [voir Utilisation dans des populations spécifiques ]. Il n'y a eu aucun résultat lié au médicament chez les mères. Informez les femmes enceintes du risque potentiel de préjudice pour le fœtus.
Le risque de fond estimé de malformations congénitales majeures et de fausse couche pour la population indiquée est inconnu. Toutes les grossesses ont un risque de fond de malformation congénitale, de perte ou d'autres résultats indésirables. Dans la population générale des États-Unis, le risque de fond estimé de malformations congénitales majeures et de fausse couche dans les grossesses cliniquement reconnues est de 2 à 4 % et de 15 à 20 %, respectivement.
Données
Données humaines
Les données disponibles provenant d'études observationnelles publiées n'ont pas réussi à démontrer une association entre les résultats indésirables liés à la grossesse et l'utilisation du pantoprazole. Les limites méthodologiques de ces études observationnelles ne permettent pas d'établir ou d'exclure définitivement tout risque médicamenteux pendant la grossesse. Dans une étude prospective menée par le Réseau européen des services d'information sur la tératologie, les résultats d'un groupe de 53 femmes enceintes ayant reçu des doses quotidiennes médianes de 40 mg de pantoprazole ont été comparés à un groupe témoin de 868 femmes enceintes qui n'ont pris aucun inhibiteur de la pompe à protons (IPP) . Il n'y avait pas de différence dans le taux de malformations majeures entre les femmes exposées aux IPP et le groupe témoin, correspondant à un risque relatif (RR) = 0,55, [intervalle de confiance (IC) à 95 % 0,08-3,95]. Dans une étude de cohorte rétrospective basée sur la population couvrant toutes les naissances vivantes au Danemark de 1996 à 2008, il n'y a pas eu d'augmentation significative des malformations congénitales majeures lors de l'analyse de l'exposition au pantoprazole au cours du premier trimestre chez 549 naissances vivantes. Une méta-analyse qui a comparé 1 530 femmes enceintes exposées aux IPP au moins au cours du premier trimestre avec 133 410 femmes enceintes non exposées n'a montré aucune augmentation significative du risque de malformations congénitales ou d'avortement spontané avec l'exposition aux IPP (pour les malformations majeures OR = 1,12 ([95 % IC 0,86-1,45] et pour les avortements spontanés OR=1,29 [IC 95% 0,84-1,97]).
Données animales
Des études de reproduction ont été réalisées chez le rat à des doses orales de pantoprazole allant jusqu'à 450 mg/kg/jour (environ 88 fois la dose humaine recommandée en fonction de la surface corporelle) et chez le lapin à des doses orales allant jusqu'à 40 mg/kg/jour (environ 16 fois la dose humaine recommandée basée sur la surface corporelle) avec l'administration de pantoprazole sodique pendant l'organogenèse chez les animaux gravides. Les études n'ont révélé aucune preuve d'altération de la fertilité ou d'atteinte au fœtus due au pantoprazole.
Une étude de toxicité sur le développement prénatal et postnatal chez le rat avec des critères d'évaluation supplémentaires pour évaluer l'effet sur le développement osseux a été réalisée avec le pantoprazole sodique. Des doses orales de pantoprazole de 5, 15 et 30 mg/kg/jour (environ 1, 3 et 6 fois la dose humaine de 40 mg/jour sur la base de la surface corporelle) ont été administrées à des femmes enceintes à partir du jour de la gestation (JG) 6 au jour de lactation (LD) 21. Du jour postnatal (PND 4) au PND 21, les chiots ont reçu des doses orales de 5, 15 et 30 mg/kg/jour (environ 1, 2,3 et 3,2 fois l'exposition ( ASC) chez l'homme à la dose de 40 mg). Il n'y a eu aucun résultat lié au médicament chez les mères. Au cours de la phase de dosage présevrage (PND 4 à 21) des ratons, il y a eu une augmentation de la mortalité et/ou de la moribondité et une diminution du poids corporel et du gain de poids corporel à 5 mg/kg/jour (expositions à peu près égales (ASC) chez les humains recevant les 40 mg) et des doses plus élevées. Au JPN 21, une diminution de la longueur et du poids moyens du fémur et des modifications de la masse osseuse et de la géométrie du fémur ont été observées chez la progéniture à 5 mg/kg/jour (expositions à peu près égales (ASC) chez l'homme à la dose de 40 mg) et à des doses plus élevées. Les résultats du fémur comprenaient une surface totale inférieure, une teneur et une densité minérales osseuses, une circonférence périostée et endostale et un moment d'inertie en coupe. Il n'y avait pas de changements microscopiques dans le fémur distal, le tibia proximal ou les articulations du grasset. Les modifications des paramètres osseux étaient partiellement réversibles après une période de récupération, les résultats au PND 70 se limitant à la densité minérale osseuse corticale/sous-corticale de la métaphyse inférieure du fémur chez les ratons femelles à 5 mg/kg/jour (expositions à peu près égales (ASC) chez l'homme à la 40 mg) et des doses plus élevées.
Lactation
Résumé des risques
Le pantoprazole a été détecté dans le lait maternel d'une mère qui allaite après une dose orale unique de 40 mg de pantoprazole. Il n'y a eu aucun effet sur le nourrisson allaité (voir Données ). Il n'y a pas de données sur les effets du pantoprazole sur la production de lait.

Les avantages de l'allaitement pour le développement et la santé doivent être pris en compte, ainsi que le besoin clinique de PROTONIX 40 mg pour la mère et tout effet indésirable potentiel du pantoprazole ou de l'affection maternelle sous-jacente sur l'enfant allaité.
Données
Le lait maternel d'une femme de 42 ans recevant 40 mg de pantoprazole par voie orale, à 10 mois post-partum, a été étudié pendant 24 heures, pour démontrer de faibles niveaux de pantoprazole présents dans le lait maternel. Le pantoprazole n'était détectable dans le lait que 2 et 4 heures après l'administration de la dose avec des concentrations dans le lait d'environ 36 mcg/L et 24 mcg/L, respectivement. Un rapport lait/plasma de 0,022 a été observé 2 heures après l'administration du médicament. Le pantoprazole n'était pas détectable (
Utilisation pédiatrique
L'innocuité et l'efficacité de PROTONIX pour le traitement à court terme (jusqu'à huit semaines) de l'EE associée au RGO ont été établies chez les patients pédiatriques âgés de 1 an à 16 ans. L'efficacité pour l'EE n'a pas été démontrée chez les patients de moins de 1 an. De plus, pour les patients de moins de 5 ans, il n'existe pas de dosage approprié dans une formulation adaptée à l'âge disponible. Par conséquent, PROTONIX est indiqué pour le traitement à court terme de l'EE associée au RGO chez les patients de 5 ans et plus. La sécurité et l'efficacité de PROTONIX pour des utilisations pédiatriques autres que l'EE n'ont pas été établies.
De 1 an à 16 ans
L'utilisation de PROTONIX chez les patients pédiatriques âgés de 1 an à 16 ans pour le traitement à court terme (jusqu'à huit semaines) de l'EE associée au RGO est étayée par : a) l'extrapolation des résultats d'études adéquates et bien contrôlées qui ont appuyé l'approbation de PROTONIX pour le traitement de l'EE associé au RGO chez l'adulte, et b) des études d'innocuité, d'efficacité et de pharmacocinétique réalisées chez des patients pédiatriques [voir Etudes cliniques , PHARMACOLOGIE CLINIQUE ].
L'innocuité de PROTONIX 40 mg dans le traitement de l'EE associée au RGO chez les patients pédiatriques âgés de 1 à 16 ans a été évaluée dans trois études multicentriques, randomisées, à double insu et à traitement parallèle, impliquant 249 patients pédiatriques, dont 8 atteints d'EE (4 patients âgés de 1 an à 5 ans et 4 patients de 5 ans à 11 ans). Les enfants âgés de 1 à 5 ans atteints d'EE diagnostiquée par endoscopie (défini comme un score Hetzel-Dent endoscopique ≥ 2) ont été traités une fois par jour pendant 8 semaines avec l'un des deux niveaux de dose de PROTONIX (environ 0,6 mg/kg ou 1,2 mg/kg ). Tous les 4 de ces patients avec EE ont été guéris (score de Hetzel-Dent de 0 ou 1) à 8 semaines. Étant donné que l'EE est peu fréquente dans la population pédiatrique, des patients principalement pédiatriques atteints de RGO symptomatique ou prouvé par endoscopie ont également été inclus dans ces études. Les patients ont été traités avec une gamme de doses de PROTONIX 20 mg une fois par jour pendant 8 semaines. Pour les résultats de sécurité, voir EFFETS INDÉSIRABLES Étant donné que ces essais pédiatriques n'avaient pas de placebo, de comparateur actif ou de preuve d'une réponse à la dose, les essais n'étaient pas concluants concernant le bénéfice clinique de PROTONIX 20 mg pour le RGO symptomatique dans la population pédiatrique. L'efficacité de PROTONIX 40 mg pour le traitement du RGO symptomatique chez les patients pédiatriques n'a pas été établie.
Bien que les données des essais cliniques soutiennent l'utilisation de PROTONIX pour le traitement à court terme de l'EE associé au RGO chez les patients pédiatriques de 1 à 5 ans, il n'existe pas de formulation posologique disponible dans le commerce appropriée pour les patients de moins de 5 ans [voir DOSAGE ET ADMINISTRATION ].
Dans une analyse pharmacocinétique de population, les valeurs de clairance chez les enfants de 1 à 5 ans atteints de RGO confirmé par endoscopie avaient une valeur médiane de 2,4 L/h. Après une dose équivalente de 1,2 mg/kg (15 mg pour ≤ 12,5 kg et 20 mg pour > 12,5 à
Nouveau-nés à moins d'un an
PROTONIX 20 mg ne s'est pas avéré efficace dans une étude multicentrique, randomisée, en double aveugle, contrôlée par placebo, sur l'arrêt du traitement de 129 patients pédiatriques âgés de 1 à 11 mois. Les patients étaient inscrits s'ils avaient un RGO symptomatique sur la base des antécédents médicaux et n'avaient pas répondu aux interventions non pharmacologiques pour le RGO pendant deux semaines. Les patients ont reçu PROTONIX quotidiennement pendant quatre semaines dans une phase en ouvert, puis les patients ont été randomisés à parts égales pour recevoir le traitement PROTONIX 40 mg ou un placebo pendant les quatre semaines suivantes en double aveugle. L'efficacité a été évaluée en observant le temps écoulé entre la randomisation et l'arrêt de l'étude en raison de l'aggravation des symptômes au cours de la phase d'arrêt du traitement de quatre semaines. Il n'y avait pas de différence statistiquement significative entre PROTONIX 20 mg et le placebo dans le taux d'arrêt.
Dans cet essai, les effets indésirables les plus fréquemment rapportés (différence ≥ 4 %) dans la population traitée par rapport à la population placebo étaient une élévation de la CK, une otite moyenne, une rhinite et une laryngite.
Dans une analyse pharmacocinétique de population, l'exposition systémique était plus élevée chez les patients de moins de 1 an atteints de RGO par rapport aux adultes ayant reçu une dose unique de 40 mg (la moyenne géométrique de l'ASC était de 103 % plus élevée chez les nourrissons prématurés et les nouveau-nés recevant une dose unique de 2,5 mg de PROTONIX et 23 % plus élevé chez les nourrissons âgés de 1 à 11 mois recevant une dose unique d'environ 1,2 mg/kg). Chez ces patients, la clairance apparente (CL/F) a augmenté avec l'âge (clairance médiane : 0,6 L/h, intervalle : 0,03 à 3,2 L/h).
Ces doses ont entraîné des effets pharmacodynamiques sur le pH gastrique mais pas sur l'œsophage. Après l'administration une fois par jour de 2,5 mg de PROTONIX 20 mg chez les prématurés et les nouveau-nés, il y a eu une augmentation du pH gastrique moyen (de 4,3 au départ à 5,2 à l'état d'équilibre) et du pourcentage moyen de temps pendant lequel le pH gastrique était > 4 ( de 60 % au départ à 80 % à l'état d'équilibre). Après une administration une fois par jour d'environ 1,2 mg/kg de PROTONIX 20 mg chez les nourrissons âgés de 1 à 11 mois, il y a eu une augmentation du pH gastrique moyen (de 3,1 au départ à 4,2 à l'état d'équilibre) et du pourcentage moyen de le pH gastrique était > 4 (de 32 % au départ à 60 % à l'état d'équilibre). Cependant, aucun changement significatif n'a été observé dans le pH intra-œsophagien moyen ou le pourcentage de temps pendant lequel le pH œsophagien était
Étant donné que PROTONIX 20 mg ne s'est pas avéré efficace dans l'étude randomisée contrôlée par placebo dans ce groupe d'âge, l'utilisation de PROTONIX pour le traitement du RGO symptomatique chez les nourrissons de moins de 1 an n'est pas indiquée.
Données sur la toxicité animale
Dans une étude sur le développement prénatal et postnatal chez le rat, les ratons ont reçu des doses orales de pantoprazole de 5, 15 et 30 mg/kg/jour (environ 1, 2,3 et 3,2 fois l'exposition (ASC) chez les enfants âgés 6 à 11 ans à une dose de 40 mg) du jour postnatal (PND 4) à PND 21, en plus de l'exposition pendant la lactation par le lait. Au JPN 21, une diminution de la longueur et du poids moyens du fémur et des modifications de la masse osseuse et de la géométrie du fémur ont été observées chez la progéniture à 5 mg/kg/jour (expositions approximativement égales (ASC) chez les enfants âgés de 6 à 11 ans à la dose de 40 mg) et des doses plus élevées. Les modifications des paramètres osseux étaient partiellement réversibles après une période de récupération.
Chez les animaux nouveau-nés/juvéniles (rats et chiens), les toxicités étaient similaires à celles observées chez les animaux adultes, notamment des altérations gastriques, une diminution de la masse des globules rouges, une augmentation des lipides, une induction enzymatique et une hypertrophie hépatocellulaire. Une incidence accrue de cellules principales éosinophiles chez les rats adultes et nouveau-nés/juvéniles, et une atrophie des cellules principales chez les rats adultes et chez les chiens nouveau-nés/juvéniles, ont été observées dans la muqueuse fundique de l'estomac dans des études à doses répétées. Une récupération complète à partielle de ces effets a été notée chez les animaux des deux groupes d'âge après une période de récupération.
Utilisation gériatrique
Dans des essais cliniques américains à court terme, les taux de guérison de l'EE chez les 107 patients âgés (≥ 65 ans) traités avec PROTONIX étaient similaires à ceux observés chez les patients de moins de 65 ans. Les taux d'incidence des effets indésirables et des anomalies de laboratoire chez les patients âgés 65 ans et plus étaient similaires à ceux associés aux patients de moins de 65 ans.
SURDOSAGE
L'expérience chez les patients prenant de très fortes doses de PROTONIX (supérieures à 240 mg) est limitée. Les rapports post-commercialisation spontanés de surdosage se situent généralement dans le profil d'innocuité connu de PROTONIX.
Le pantoprazole n'est pas éliminé par hémodialyse. En cas de surdosage, le traitement doit être symptomatique et de soutien.
Des doses orales uniques de pantoprazole à 709 mg/kg, 798 mg/kg et 887 mg/kg ont été mortelles pour les souris, les rats et les chiens, respectivement. Les symptômes de toxicité aiguë étaient l'hypoactivité, l'ataxie, la position courbée, l'écartement des membres, la position latérale, la ségrégation, l'absence de réflexe auriculaire et les tremblements.
En cas de surexposition à PROTONIX 20 mg, appelez votre centre antipoison au 1-800-222-1222 pour obtenir des informations à jour sur la prise en charge d'un empoisonnement ou d'un surdosage.
CONTRE-INDICATIONS
- PROTONIX est contre-indiqué chez les patients présentant une hypersensibilité connue à l'un des composants de la formulation ou à tout benzimidazole substitué. Les réactions d'hypersensibilité peuvent inclure l'anaphylaxie, le choc anaphylactique, l'œdème de Quincke, le bronchospasme, la néphrite tubulo-interstitielle aiguë et l'urticaire [voir AVERTISSEMENTS ET PRECAUTIONS , EFFETS INDÉSIRABLES ].
- Les inhibiteurs de la pompe à protons (IPP), y compris PROTONIX, sont contre-indiqués chez les patients recevant des produits contenant de la rilpivirine [voir INTERACTIONS MÉDICAMENTEUSES ].
PHARMACOLOGIE CLINIQUE
Mécanisme d'action
Le pantoprazole est un IPP qui supprime l'étape finale de la production d'acide gastrique en se liant de manière covalente au système enzymatique (H+, K+)-ATPase à la surface sécrétoire de la cellule pariétale gastrique. Cet effet conduit à l'inhibition de la sécrétion basale et stimulée d'acide gastrique, quel que soit le stimulus. La liaison à la (H+, K+)-ATPase se traduit par une durée d'effet antisécrétoire qui persiste au-delà de 24 heures pour toutes les doses testées (20 mg à 120 mg).
Pharmacodynamie
Il a été démontré que PROTONIX 20 mg pour suspension orale à libération retardée, 40 mg, est comparable aux comprimés à libération retardée PROTONIX dans la suppression de la MAO stimulée par la pentagastrine chez les patients (n = 49) atteints de RGO et ayant des antécédents d'EE. Dans cette étude pharmacodynamique croisée multicentrique, une dose orale de 40 mg de PROTONIX pour suspension orale à libération retardée administrée dans une cuillerée à thé de compote de pommes a été comparée à une dose orale de 40 mg de comprimés à libération retardée PROTONIX après l'administration de chaque formulation une fois par jour pendant 7 journées. Les deux médicaments ont été administrés trente minutes avant le petit déjeuner. Stimulé par la pentagastrine (MAO) a été évalué de l'heure 23 à 24 à l'état d'équilibre.
Activité antisécrétoire
Dans des conditions de stimulation acide maximales utilisant la pentagastrine, une diminution dose-dépendante du débit d'acide gastrique se produit après une dose unique de pantoprazole oral (20-80 mg) ou intraveineux (20-120 mg) chez des sujets sains. Le pantoprazole administré une fois par jour entraîne une augmentation de l'inhibition de la sécrétion d'acide gastrique. Après la dose orale initiale de 40 mg de pantoprazole, une inhibition moyenne de 51 % a été obtenue en 2,5 heures. Avec une administration une fois par jour pendant 7 jours, l'inhibition moyenne a été augmentée à 85 %. Le pantoprazole a supprimé la sécrétion acide à plus de 95 % chez la moitié des sujets. La sécrétion acide était revenue à la normale dans la semaine suivant la dernière dose de pantoprazole ; il n'y avait aucune preuve d'hypersécrétion rebond.
Dans une série d'études dose-réponse, le pantoprazole, à des doses orales allant de 20 à 120 mg, a provoqué des augmentations liées à la dose du pH gastrique basal médian et du pourcentage de fois où le pH gastrique était > 3 et > 4. Le traitement avec 40 mg de pantoprazole a produit des augmentations significativement plus importantes du pH gastrique que la dose de 20 mg. Des doses supérieures à 40 mg (60, 80, 120 mg) n'ont pas entraîné d'autres augmentations significatives du pH gastrique médian. Les effets du pantoprazole sur le pH médian d'une étude croisée en double aveugle sont présentés dans le tableau 5.
Effets de la gastrine sérique
Les taux de gastrine sérique à jeun ont été évalués dans deux études en double aveugle sur la guérison aiguë de l'EE dans lesquelles 682 patients atteints de reflux gastro-œsophagien (RGO) ont reçu 10, 20 ou 40 mg de PROTONIX 20 mg pendant jusqu'à 8 semaines. À 4 semaines de traitement, il y a eu une augmentation des taux moyens de gastrine de 7 %, 35 % et 72 % par rapport aux valeurs de prétraitement dans les groupes de traitement de 10, 20 et 40 mg, respectivement. Une augmentation similaire des taux sériques de gastrine a été notée lors de la visite de 8 semaines avec des augmentations moyennes de 3 %, 26 % et 84 % pour les trois groupes de doses de pantoprazole. Les taux sériques médians de gastrine sont restés dans les limites normales pendant le traitement d'entretien avec les comprimés à libération retardée PROTONIX.
Dans des études internationales à long terme portant sur plus de 800 patients, une augmentation moyenne de 2 à 3 fois par rapport au niveau de gastrine sérique à jeun avant le traitement a été observée au cours des premiers mois de traitement par le pantoprazole à des doses de 40 mg par jour pendant les études d'entretien du RGO et 40 mg ou plus par jour chez les patients atteints de RGO réfractaire. Les taux sériques de gastrine à jeun sont généralement restés à environ 2 à 3 fois le niveau initial pendant jusqu'à 4 ans de suivi périodique dans les essais cliniques.
Après un traitement à court terme avec PROTONIX 20 mg, les taux élevés de gastrine reviennent à la normale en au moins 3 mois.
Effets sur les cellules de type entérochromaffine (ECL)
Chez 39 patients traités par pantoprazole oral 40 mg à 240 mg par jour (la majorité recevant 40 mg à 80 mg) pendant une période allant jusqu'à 5 ans, il y a eu une augmentation modérée de la densité des cellules ECL, commençant après la première année d'utilisation, qui a semblé plateau après 4 ans.
Dans une étude non clinique chez des rats Sprague-Dawley, une exposition à vie (24 mois) au pantoprazole à des doses de 0,5 à 200 mg/kg/jour a entraîné une augmentation liée à la dose de la prolifération des cellules ECL gastriques et des tumeurs des cellules neuroendocrines (NE) gastriques. . Les tumeurs des cellules NE gastriques chez le rat peuvent résulter d'une élévation chronique des concentrations sériques de gastrine. La forte densité de cellules ECL dans l'estomac du rat rend cette espèce très sensible aux effets prolifératifs des concentrations élevées de gastrine produites par les IPP. Cependant, aucune élévation de la gastrine sérique n'a été observée après l'administration de pantoprazole à une dose de 0,5 mg/kg/jour. Dans une étude distincte, une tumeur des cellules NE gastriques sans modifications concomitantes de la prolifération des cellules ECL a été observée chez 1 rat femelle après 12 mois d'administration de pantoprazole à 5 mg/kg/jour et 9 mois de récupération après l'arrêt de la dose [voir Toxicologie non clinique ].
Effets endocriniens
Dans une étude de pharmacologie clinique, PROTONIX 40 mg administré une fois par jour pendant 2 semaines n'a eu aucun effet sur les taux des hormones suivantes : cortisol, testostérone, triiodothyronine (T3), thyroxine (T4), thyréostimuline (TSH), thyronine- protéine de liaison, hormone parathyroïdienne, insuline, glucagon, rénine, aldostérone, hormone folliculo-stimulante, hormone lutéinisante, prolactine et hormone de croissance.
Dans une étude d'un an sur des patients atteints de RGO traités par PROTONIX 40 mg ou 20 mg, il n'y a eu aucun changement par rapport au départ dans les niveaux globaux de T3, T4 et TSH.
Pharmacocinétique
Les comprimés à libération retardée PROTONIX 40 mg sont préparés sous forme de comprimés à enrobage entérique de sorte que l'absorption du pantoprazole ne commence qu'après que le comprimé a quitté l'estomac. La concentration sérique maximale (Cmax) et l'aire sous la courbe de concentration sérique en fonction du temps (ASC) augmentent de manière proportionnelle aux doses orales et intraveineuses de 10 mg à 80 mg. Le pantoprazole ne s'accumule pas et sa pharmacocinétique n'est pas modifiée en cas d'administration quotidienne multiple. Après administration orale ou intraveineuse, la concentration sérique de pantoprazole diminue de façon biexponentielle, avec une demi-vie d'élimination terminale d'environ une heure.
Chez les métaboliseurs rapides ayant une fonction hépatique normale recevant une dose orale du comprimé de pantoprazole à enrobage entérique de 40 mg, la concentration maximale (Cmax) est de 2,5 μg/mL ; le temps nécessaire pour atteindre la concentration maximale (tmax) est de 2,5 h, et l'aire totale moyenne sous la courbe de concentration plasmatique en fonction du temps (AUC) est de 4,8 μg h/mL (plage de 1,4 à 13,3 μg h/mL). Après administration intraveineuse de pantoprazole à des métaboliseurs rapides, sa clairance totale est de 7,6 à 14,0 L/h et son volume apparent de distribution est de 11,0 à 23,6 L.
Une dose orale unique de PROTONIX pour suspension orale à libération retardée, 40 mg, s'est avérée bioéquivalente lorsqu'elle est administrée à des sujets sains (N = 22) sous forme de granulés saupoudrés sur une cuillerée à thé de compote de pommes, sous forme de granulés mélangés à du jus de pomme ou mélangés à jus de pomme suivi d'une administration par sonde nasogastrique. Les paramètres pharmacocinétiques plasmatiques d'une étude croisée chez des sujets sains sont résumés dans le tableau 6.
Absorption
Après l'administration d'une ou plusieurs doses orales de 40 mg de comprimés à libération retardée de PROTONIX 20 mg, la concentration plasmatique maximale de pantoprazole a été atteinte en environ 2,5 heures et la Cmax était de 2,5 μg/mL. Le pantoprazole subit peu de métabolisme de premier passage, ce qui entraîne une biodisponibilité absolue d'environ 77 %. L'absorption du pantoprazole n'est pas affectée par l'administration concomitante d'antiacides.
L'administration de PROTONIX comprimés à libération retardée avec de la nourriture peut retarder son absorption jusqu'à 2 heures ou plus ; cependant, la Cmax et le degré d'absorption du pantoprazole (AUC) ne sont pas modifiés. Ainsi, les comprimés à libération retardée PROTONIX 40 mg peuvent être pris sans tenir compte de l'heure des repas.
L'administration de granules de pantoprazole, 40 mg, avec un repas riche en graisses a retardé le temps médian jusqu'au pic de concentration plasmatique de 2 heures. Avec un repas concomitant riche en graisses, la Cmax et l'ASC des granules de pantoprazole, 40 mg, saupoudrés sur de la compote de pommes ont diminué de 51 % et 29 %, respectivement. Ainsi, PROTONIX 20 mg pour suspension buvable à libération retardée doit être pris environ 30 minutes avant un repas.
Distribution
Le volume apparent de distribution du pantoprazole est d'environ 11 à 23,6 L, distribué principalement dans le liquide extracellulaire. La liaison aux protéines sériques du pantoprazole est d'environ 98 %, principalement à l'albumine.
Élimination
Métabolisme
Le pantoprazole est largement métabolisé dans le foie par le système du cytochrome P450 (CYP). Le métabolisme du pantoprazole est indépendant de la voie d'administration (intraveineuse ou orale). La principale voie métabolique est la déméthylation, par le CYP2C19, suivie d'une sulfatation ; d'autres voies métaboliques comprennent l'oxydation par le CYP3A4. Il n'y a aucune preuve que l'un des métabolites du pantoprazole ait une activité pharmacologique significative.
Excrétion
Après une dose unique orale ou intraveineuse de pantoprazole marqué au 14C chez des sujets sains métaboliseurs normaux, environ 71 % de la dose a été excrétée dans l'urine, dont 18 % dans les fèces par excrétion biliaire. Il n'y a pas eu d'excrétion rénale du pantoprazole inchangé.
Populations spécifiques
Patients gériatriques
Seules des augmentations légères à modérées de l'ASC (43 %) et de la Cmax (26 %) du pantoprazole ont été observées chez les sujets âgés (64 à 76 ans) après administration orale répétée, par rapport aux sujets plus jeunes [voir Utilisation dans des populations spécifiques ].
Patients pédiatriques
La pharmacocinétique du pantoprazole a été étudiée chez des enfants de moins de 16 ans dans quatre essais cliniques randomisés en ouvert chez des patients pédiatriques atteints de RGO présumé/avéré. Une formulation pédiatrique de granulés a été étudiée chez des enfants jusqu'à 5 ans, et les comprimés à libération retardée de PROTONIX 40 mg ont été étudiés chez des enfants de plus de 5 ans.
Dans une analyse pharmacocinétique de population, la clairance totale a augmenté avec l'augmentation du poids corporel de manière non linéaire. La clairance totale a également augmenté avec l'âge uniquement chez les enfants de moins de 3 ans.
Nouveau-né jusqu'à 5 ans
[voir Utilisation dans des populations spécifiques ]
Enfants et adolescents de 6 à 16 ans
La pharmacocinétique des comprimés à libération retardée PROTONIX a été évaluée chez des enfants âgés de 6 à 16 ans avec un diagnostic clinique de RGO. Les paramètres pharmacocinétiques après une dose orale unique de 20 mg ou 40 mg de comprimés PROTONIX 40 mg chez les enfants âgés de 6 à 16 ans étaient très variables (% CV allant de 40 à 80 %). La moyenne géométrique de l'ASC estimée à partir de l'analyse pharmacocinétique de population après un comprimé de 40 mg de PROTONIX à 40 mg chez les patients pédiatriques était d'environ 39 % et 10 % plus élevée respectivement chez les enfants de 6 à 11 ans et de 12 à 16 ans, par rapport à celle des adultes (Tableau 7) .
Patients masculins et féminins
Il y a une légère augmentation de l'ASC et de la Cmax du pantoprazole chez les femmes par rapport aux hommes. Cependant, les valeurs de clairance normalisées en fonction du poids sont similaires chez les femmes et les hommes.
Chez les patients pédiatriques âgés de 1 à 16 ans, il n'y a eu aucun effet cliniquement pertinent du sexe sur la clairance du pantoprazole, comme le montre l'analyse pharmacocinétique de population.
Patients atteints d'insuffisance rénale
Chez les patients atteints d'insuffisance rénale sévère, les paramètres pharmacocinétiques du pantoprazole étaient similaires à ceux des sujets sains.
Patients atteints d'insuffisance hépatique
Chez les patients atteints d'insuffisance hépatique légère à sévère (cirrhose de Child-Pugh A à C), les concentrations maximales de pantoprazole n'ont augmenté que légèrement (1,5 fois) par rapport aux sujets sains. Bien que les valeurs de demi-vie sérique aient augmenté de 7 à 9 heures et que les valeurs d'ASC aient été multipliées par 5 à 7 chez les patients insuffisants hépatiques, ces augmentations n'étaient pas supérieures à celles observées chez les métaboliseurs lents du CYP2C19, pour lesquels aucun ajustement posologique n'est justifié. Ces changements pharmacocinétiques chez les patients insuffisants hépatiques entraînent une accumulation minimale de médicament après l'administration de doses multiples une fois par jour. Des doses supérieures à 40 mg/jour n'ont pas été étudiées chez les insuffisants hépatiques.
Études sur les interactions médicamenteuses
Effet d'autres médicaments sur le pantoprazole
Le pantoprazole est métabolisé principalement par le CYP2C19 et, dans une moindre mesure, par les CYP 3A4, 2D6 et 2C9. Dans des études in vivo sur les interactions médicamenteuses avec des substrats du CYP2C19 (diazépam [également un substrat du CYP3A4] et phénytoïne [également un inducteur du CYP3A4] et clopidogrel), la nifédipine, le midazolam et la clarithromycine (substrats du CYP3A4), le métoprolol (un substrat du CYP2D6), le diclofénac , le naproxène et le piroxicam (substrats du CYP2C9) et la théophylline (un substrat du CYP1A2) chez des sujets sains, la pharmacocinétique du pantoprazole n'a pas été modifiée de manière significative.
Effet du pantoprazole sur d'autres médicaments
Clopidogrel
Le clopidogrel est métabolisé en son métabolite actif en partie par le CYP2C19. Dans une étude clinique croisée, 66 sujets sains ont reçu du clopidogrel (dose de charge de 300 mg suivie de 75 mg par jour) seul et avec du pantoprazole (80 mg en même temps que le clopidogrel) pendant 5 jours. Au jour 5, l'ASC moyenne du métabolite actif du clopidogrel a été réduite d'environ 14 % (le rapport moyen géométrique était de 86 %, avec un IC à 90 % de 79 à 93 %) lorsque le pantoprazole était co-administré avec le clopidogrel par rapport au clopidogrel administré seul. Des paramètres pharmacodynamiques ont également été mesurés et ont démontré que la modification de l'inhibition de l'agrégation plaquettaire (induite par l'ADP 5 μM) était corrélée à la modification de l'exposition au métabolite actif du clopidogrel. La signification clinique de cette découverte n'est pas claire.
Mycophénolate mofétil (MMF)
L'administration de pantoprazole 40 mg deux fois par jour pendant 4 jours et d'une dose unique de 1000 mg de MMF environ une heure après la dernière dose de pantoprazole à 12 sujets sains dans une étude croisée a entraîné une réduction de 57 % de la Cmax et une réduction de 27 % dans l'AUC de l'AMP. Des patients transplantés recevant environ 2000 mg par jour de MMF (n = 12) ont été comparés à des patients transplantés recevant environ la même dose de MMF et de pantoprazole 40 mg par jour (n = 21). Il y a eu une réduction de 78 % de la Cmax et une réduction de 45 % de l'ASC du MPA chez les patients recevant à la fois du pantoprazole et du MMF [voir INTERACTIONS MÉDICAMENTEUSES ].
Autres drogues
Des études in vivo suggèrent également que le pantoprazole n'affecte pas significativement la cinétique des médicaments suivants (cisapride, théophylline, diazépam [et son métabolite actif, le déméthyldiazépam], phénytoïne, métoprolol, nifédipine, carbamazépine, midazolam, clarithromycine, diclofénac, naproxène, piroxicam, et contraceptifs oraux [lévonorgestrel/éthinylestradiol]). Dans d'autres études in vivo, la digoxine, l'éthanol, le glyburide, l'antipyrine, la caféine, le métronidazole et l'amoxicilline n'ont eu aucune interaction cliniquement pertinente avec le pantoprazole.
Bien qu'aucune interaction médicamenteuse significative n'ait été observée dans les études cliniques, le potentiel d'interactions médicamenteuses significatives avec des doses élevées de pantoprazole plus d'une fois par jour n'a pas été étudié chez les métaboliseurs lents ou chez les personnes atteintes d'insuffisance hépatique.
Antiacides
Il n'y a pas non plus eu d'interaction avec les antiacides administrés en concomitance.
Pharmacogénomique
Le CYP2C19 présente un polymorphisme génétique connu en raison de son déficit dans certaines sous-populations (par exemple, environ 3 % des Caucasiens et des Afro-Américains et 17 % à 23 % des Asiatiques sont des métaboliseurs lents). Bien que ces sous-populations de métaboliseurs lents du pantoprazole aient des valeurs de demi-vie d'élimination de 3,5 à 10 heures chez l'adulte, elles présentent toujours une accumulation minimale (23 % ou moins) avec une administration uniquotidienne. Pour les patients adultes métaboliseurs lents du CYP2C19, aucun ajustement posologique n'est nécessaire.
Comme pour les adultes, les patients pédiatriques qui ont le génotype de métaboliseur lent du CYP2C19 (CYP2C19 *2/*2) ont présenté une augmentation de plus de 6 fois de l'ASC par rapport aux patients pédiatriques étendus (CYP2C19 *1/*1) et intermédiaires (CYP2C19 *1 /*x) métaboliseurs. Les métaboliseurs lents présentaient une clairance orale apparente environ 10 fois inférieure à celle des métaboliseurs rapides.
Pour les métaboliseurs lents pédiatriques connus, une réduction de la dose doit être envisagée.
Etudes cliniques
Les comprimés à libération retardée PROTONIX 20 mg ont été utilisés dans les essais cliniques suivants.
Oesophagite érosive (EE) associée au reflux gastro-oesophagien (RGO)
Patients adultes
Une étude américaine multicentrique, en double aveugle et contrôlée par placebo de PROTONIX 10 mg, 20 mg ou 40 mg une fois par jour a été menée chez 603 patients présentant des symptômes de reflux et une EE diagnostiquée par endoscopie de grade 2 ou plus (échelle de Hetzel-Dent). Dans cette étude, environ 25 % des patients inclus avaient une EE sévère de grade 3 et 10 % avaient un grade 4. Les pourcentages de patients guéris (selon le protocole, n = 541) dans cette étude sont présentés dans le tableau 8.
Dans cette étude, tous les groupes de traitement PROTONIX avaient des taux de guérison significativement plus élevés que le groupe placebo. Cela était vrai quel que soit le statut H. pylori pour les groupes de traitement 40 mg et 20 mg PROTONIX 40 mg. La dose de 40 mg de PROTONIX 20 mg a entraîné des taux de guérison significativement supérieurs à ceux observés avec la dose de 20 mg ou de 10 mg.
Une proportion significativement plus élevée de patients prenant PROTONIX 40 mg a présenté un soulagement complet des brûlures d'estomac diurnes et nocturnes et l'absence de régurgitation, dès le premier jour de traitement, par rapport au placebo. Les patients prenant PROTONIX ont consommé beaucoup moins de comprimés antiacides par jour que ceux prenant un placebo.
PROTONIX 40 mg et 20 mg une fois par jour ont également été comparés à la nizatidine 150 mg deux fois par jour dans une étude américaine multicentrique en double aveugle portant sur 243 patients présentant des symptômes de reflux et une EE de grade 2 ou plus diagnostiquée par endoscopie. Les pourcentages de patients guéris (selon le protocole, n = 212) sont présentés dans le tableau 9.
Le traitement une fois par jour avec PROTONIX 40 mg ou 20 mg a entraîné des taux de guérison significativement supérieurs à 4 et 8 semaines par rapport au traitement deux fois par jour avec 150 mg de nizatidine. Pour le groupe de traitement de 40 mg, des taux de guérison significativement plus élevés par rapport à la nizatidine ont été obtenus quel que soit le statut H. pylori.
Une proportion significativement plus élevée de patients dans les groupes de traitement PROTONIX ont ressenti un soulagement complet des brûlures d'estomac et des régurgitations nocturnes, à partir du premier jour, et des brûlures d'estomac diurnes le deuxième jour, par rapport à ceux prenant de la nizatidine 150 mg deux fois par jour. Les patients prenant PROTONIX ont consommé beaucoup moins de comprimés antiacides par jour que ceux prenant de la nizatidine.
Patients pédiatriques âgés de 5 à 16 ans
L'efficacité de PROTONIX dans le traitement de l'EE associée au RGO chez les patients pédiatriques âgés de 5 à 16 ans est extrapolée à partir d'essais adéquats et bien menés chez l'adulte, car on pense que la physiopathologie est la même. Quatre patients pédiatriques atteints d'EE diagnostiquée par endoscopie ont été étudiés dans des essais multicentriques, randomisés, en double aveugle et à traitement parallèle. Les enfants atteints d'EE diagnostiquée par endoscopie (défini comme un score Hetzel-Dent endoscopique ≥ 2) ont été traités une fois par jour pendant 8 semaines avec l'un des deux niveaux de dose de PROTONIX (20 mg ou 40 mg). Les 4 patients atteints d'EE étaient guéris (score Hetzel-Dent de 0 ou 1) à 8 semaines.
Maintien à long terme de la guérison de l'œsophagite érosive
Deux essais indépendants, multicentriques, randomisés, en double aveugle et contrôlés par comparateur de conception identique ont été menés chez des patients adultes atteints de RGO avec une EE cicatrisée confirmée par endoscopie pour démontrer l'efficacité de PROTONIX dans le maintien à long terme de la cicatrisation. Les deux études américaines ont recruté 386 et 404 patients, respectivement, pour recevoir soit 10 mg, 20 mg ou 40 mg de PROTONIX 40 mg comprimés à libération retardée une fois par jour ou 150 mg de ranitidine deux fois par jour. Comme démontré dans le tableau 10, PROTONIX 40 mg et 20 mg étaient significativement supérieurs à la ranitidine à chaque instant en ce qui concerne le maintien de la cicatrisation. De plus, PROTONIX 40 mg a été supérieur à tous les autres traitements étudiés.
PROTONIX 40 mg a été supérieur à la ranitidine pour réduire le nombre d'épisodes de brûlures d'estomac diurnes et nocturnes du premier au douzième mois de traitement. PROTONIX 20 mg, administré une fois par jour, a également été efficace pour réduire les épisodes de brûlures d'estomac diurnes et nocturnes dans un essai, comme présenté dans le tableau 11.
Conditions hypersécrétoires pathologiques, y compris le syndrome de Zollinger-Ellison
Dans un essai ouvert multicentrique portant sur 35 patients atteints d'hypersécrétions pathologiques, telles que le syndrome de Zollinger-Ellison, avec ou sans néoplasie endocrinienne multiple de type I, PROTONIX a contrôlé avec succès la sécrétion d'acide gastrique. Des doses allant de 80 mg par jour à 240 mg par jour ont maintenu le débit d'acide gastrique inférieur à 10 mEq/h chez les patients sans chirurgie anti-acide antérieure et inférieur à 5 mEq/h chez les patients ayant déjà subi une chirurgie anti-acide.
Les doses ont été initialement titrées en fonction des besoins individuels du patient et ajustées chez certains patients en fonction de la réponse clinique avec le temps [voir DOSAGE ET ADMINISTRATION ]. PROTONIX 40 mg a été bien toléré à ces doses pendant des périodes prolongées (plus de 2 ans chez certains patients).
INFORMATIONS PATIENTS
PROTONIX (pro-TAH-nix)(pantoprazole sodique) comprimés à libération retardée et PROTONIX (pro-TAH-nix) (pantoprazole sodique) pour suspension buvable à libération retardée
Quelles sont les informations les plus importantes que je devrais connaître sur PROTONIX ?
Vous devez prendre PROTONIX 20 mg exactement comme prescrit, à la dose la plus faible possible et pendant la durée la plus courte nécessaire.
PROTONIX peut soulager vos symptômes liés à l'acidité, mais vous pourriez tout de même avoir de graves problèmes d'estomac. Discutez avec votre médecin.
PROTONIX peut provoquer des effets secondaires graves, notamment :
- Un type de problème rénal (néphrite tubulo-interstitielle aiguë). Certaines personnes prenant des inhibiteurs de la pompe à protons (IPP), y compris PROTONIX 40 mg, peuvent développer un problème rénal appelé néphrite tubulo-interstitielle aiguë qui peut survenir à tout moment pendant le traitement par PROTONIX. Appelez immédiatement votre médecin si vous avez une diminution de la quantité que vous urinez ou si vous avez du sang dans vos urines.
- Diarrhée causée par une infection (Clostridium difficile) dans vos intestins. Appelez immédiatement votre médecin si vous avez des selles liquides ou des douleurs à l'estomac qui ne disparaissent pas. Vous pouvez ou non avoir de la fièvre.
- Fractures osseuses (hanche, poignet ou colonne vertébrale). Des fractures osseuses de la hanche, du poignet ou de la colonne vertébrale peuvent survenir chez les personnes qui prennent plusieurs doses quotidiennes d'IPP et pendant une longue période (un an ou plus). Informez votre médecin si vous avez une fracture osseuse, en particulier à la hanche, au poignet ou à la colonne vertébrale.
- Certains types de lupus érythémateux. Le lupus érythémateux est une maladie auto-immune (les cellules immunitaires du corps attaquent d'autres cellules ou organes du corps). Certaines personnes qui prennent des médicaments IPP, y compris PROTONIX 40 mg, peuvent développer certains types de lupus érythémateux ou présenter une aggravation du lupus qu'ils ont déjà. Appelez immédiatement votre médecin si vous avez des douleurs articulaires nouvelles ou qui s'aggravent ou une éruption cutanée sur les joues ou les bras qui s'aggrave au soleil.
Parlez à votre médecin de votre risque de ces effets secondaires graves.
PROTONIX peut avoir d'autres effets secondaires graves. Voir « Quels sont les effets secondaires possibles de PROTONIX ? »
Qu'est-ce que PROTONIX 20 mg ?
Un médicament sur ordonnance appelé inhibiteur de la pompe à protons (IPP) utilisé pour réduire la quantité d'acide dans votre estomac.
Chez l'adulte, PROTONIX est utilisé pour :
- jusqu'à 8 semaines pour la guérison et le soulagement des symptômes des lésions de la muqueuse de l'œsophage liées à l'acide (appelées œsophagite érosive ou EE). Votre médecin peut prescrire 8 semaines supplémentaires de PROTONIX 20 mg chez les patients dont l'EE ne guérit pas.
- maintenir la guérison de l'EE et aider à prévenir le retour des symptômes de brûlures d'estomac causés par le RGO. On ne sait pas si PROTONIX 40 mg est sûr et efficace lorsqu'il est utilisé pendant plus de 12 mois à cette fin.
- le traitement à long terme des conditions où votre estomac produit trop d'acide. Cela inclut une maladie rare appelée syndrome de Zollinger-Ellison.
Chez les enfants de 5 ans et plus, PROTONIX est utilisé pour :
- jusqu'à 8 semaines pour la guérison et le soulagement des symptômes de l'EE. On ne sait pas si PROTONIX 20 mg est sûr s'il est utilisé plus de 8 semaines chez les enfants.
PROTONIX 20 mg ne doit pas être utilisé chez les enfants de moins de 5 ans. On ne sait pas si PROTONIX 40 mg est sûr et efficace chez les enfants pour un traitement autre que l'EE.
Ne prenez jamais PROTONIX 20mg si vous êtes :
- allergique au pantoprazole sodique, à tout autre médicament IPP ou à l'un des ingrédients de PROTONIX. Voir la fin de ce Guide de Médication pour une liste complète d'ingrédients.
- prendre un médicament contenant de la rilpivirine (EDURANT, COMPLERA, ODEFSEY, JULUCA) utilisé pour traiter le VIH-1 (virus de l'immunodéficience humaine).
Avant de prendre PROTONIX, informez votre médecin de toutes vos conditions médicales, y compris si vous :
- avez un faible taux de magnésium dans le sang.
- êtes enceinte ou envisagez de devenir enceinte. PROTONIX 40 mg peut nuire à votre bébé à naître. Informez votre médecin si vous tombez enceinte ou si vous pensez être enceinte pendant le traitement par PROTONIX.
- allaitez ou envisagez d'allaiter. PROTONIX peut passer dans votre lait maternel. Discutez avec votre médecin de la meilleure façon de nourrir votre bébé si vous prenez PROTONIX.
Informez votre médecin de tous les médicaments que vous prenez, y compris les médicaments sur ordonnance et en vente libre, les vitamines et les suppléments à base de plantes. Informez surtout votre médecin si vous prenez méthotrexate (Otrexup, Rasuvo, Trexall, XATMEP), digoxine (LANOXIN) ou un diurétique (diurétique).
Comment dois-je prendre PROTONIX 20mg ?
- Prenez PROTONIX 40mg exactement comme prescrit par votre médecin.
Comprimés à libération retardée PROTONIX (comprimés PROTONIX 20 mg) :
-
- Ne pas couper, mâcher ou écraser les comprimés de PROTONIX 40 mg.
- Avalez les comprimés de PROTONIX 20mg entiers, avec ou sans nourriture.
- Informez votre médecin si vous ne parvenez pas à avaler votre comprimé de PROTONIX 20 mg.
- Vous pouvez utiliser des antiacides pendant que vous prenez les comprimés de PROTONIX 40 mg.
PROTONIX 40mg pour suspension buvable à libération retardée (PROTONIX pour suspension buvable) :
-
- Ne pas diviser, mâcher ou écraser PROTONIX 20 mg pour suspension buvable.
- Prenez PROTONIX 40 mg pour suspension buvable environ 30 minutes avant un repas.
- PROTONIX pour suspension buvable ne doit être administré que par voie orale mélangé à du jus de pomme ou de la compote de pommes ou par une sonde nasogastrique (NG) ou une sonde de gastrostomie mélangée à du jus de pomme. Ne pas mélanger PROTONIX pour suspension buvable dans des liquides autres que le jus de pomme ou des aliments autres que la compote de pommes.
- Ne divisez pas un sachet de PROTONIX 20 mg pour suspension buvable pour obtenir une dose plus petite.
- Voir le "Mode d'emploi" à la fin de ce guide des médicaments pour savoir comment mélanger et prendre PROTONIX 40 mg pour suspension buvable par voie orale dans de la compote de pommes ou du jus de pomme ou comment mélanger et administrer la suspension par un tube NG ou un tube de gastrostomie mélangé à du jus de pomme.
- Si vous manquez une dose de PROTONIX, prenez-la dès que possible. S'il est presque l'heure de votre prochaine dose, ne prenez pas la dose oubliée. Prenez la dose suivante à l'heure habituelle. Ne prenez pas 2 doses en même temps.
- Si vous avez pris trop de PROTONIX, appelez immédiatement votre médecin ou votre centre antipoison au 1-800-222-1222 ou rendez-vous à la salle d'urgence la plus proche.
Quels sont les effets secondaires possibles de PROTONIX 40mg ?
PROTONIX 20 mg peut provoquer des effets indésirables graves, notamment :
- Voir "Quelles sont les informations les plus importantes que je devrais connaître sur PROTONIX ?"
- Faibles niveaux de vitamine B-12 dans votre corps peut survenir chez les personnes qui ont pris PROTONIX 40 mg pendant une longue période (plus de 3 ans). Informez votre médecin si vous présentez des symptômes de faible taux de vitamine B-12, notamment un essoufflement, des étourdissements, un rythme cardiaque irrégulier, une faiblesse musculaire, une peau pâle, une sensation de fatigue, des changements d'humeur et des picotements ou un engourdissement dans les bras et les jambes.
- Faibles niveaux de magnésium dans votre corps peut survenir chez les personnes qui ont pris PROTONIX pendant au moins 3 mois. Informez votre médecin si vous présentez des symptômes de faibles niveaux de magnésium, notamment des convulsions, des étourdissements, un rythme cardiaque irrégulier, de la nervosité, des douleurs musculaires ou une faiblesse et des spasmes des mains, des pieds ou de la voix.
- Excroissances de l'estomac (polypes de la glande fundique). Les personnes qui prennent des médicaments IPP pendant une longue période ont un risque accru de développer un certain type d'excroissances gastriques appelées polypes de la glande fundique, en particulier après avoir pris des médicaments IPP pendant plus d'un an.
Les effets indésirables les plus courants de PROTONIX 40 mg chez l'adulte incluent : maux de tête, diarrhée, nausées, douleurs dans la région de l'estomac (abdominales), vomissements, gaz, étourdissements et douleurs articulaires.
Les effets indésirables les plus courants de PROTONIX 40 mg chez les enfants incluent : infection des voies respiratoires supérieures, maux de tête, fièvre, diarrhée, vomissements, éruption cutanée et douleurs abdominales. Ce ne sont pas tous les effets secondaires possibles de PROTONIX. Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
Comment dois-je conserver PROTONIX 40mg ?
Conservez PROTONIX 20mg à température ambiante entre 68°F et 77°F (20°C à 25°C).
Gardez PROTONIX et tous les médicaments hors de la portée des enfants.
Informations générales sur l'utilisation sûre et efficace de PROTONIX.
Les médicaments sont parfois prescrits à des fins autres que celles énumérées dans un guide des médicaments. N'utilisez pas PROTONIX pour une condition pour laquelle il n'a pas été prescrit. Ne donnez pas PROTONIX 20 mg à d'autres personnes, même si elles présentent les mêmes symptômes que vous. Cela peut leur nuire. Vous pouvez demander à votre médecin ou à votre pharmacien des informations sur PROTONIX 20 mg destinées aux professionnels de la santé.
Quels sont les ingrédients de PROTONIX ?
Ingrédient actif: pantoprazole sodique sesquihydraté
Ingrédients inactifs des comprimés à libération retardée PROTONIX : stéarate de calcium, crospovidone, hypromellose, oxyde de fer, mannitol, copolymère d'acide méthacrylique, polysorbate 80, povidone, propylène glycol, carbonate de sodium, laurylsulfate de sodium, dioxyde de titane et citrate de triéthyle.
Ingrédients inactifs de PROTONIX 40mg pour suspension buvable à libération retardée : crospovidone, hypromellose, copolymère d'acide méthacrylique, cellulose microcristalline, polysorbate 80, povidone, carbonate de sodium, laurylsulfate de sodium, talc, dioxyde de titane, citrate de triéthyle et oxyde de fer jaune.
Mode d'emploi
PROTONIX (pro-TAH-nix) (pantoprazole sodique) pour suspension buvable à libération retardée
PROTONIX 40mg pour libération retardée suspension buvable (PROTONIX 40mg pour suspension buvable) :
Une information important:
- Ne pas diviser, mâcher ou écraser PROTONIX pour suspension buvable.
- Prenez PROTONIX pour suspension buvable environ 30 minutes avant un repas.
- PROTONIX pour suspension buvable :
- devrait être pris uniquement avec de la compote de pommes ou du jus de pomme.
- devrait ne pas être mélangé dans de l'eau ou d'autres liquides, ou d'autres aliments.
- le paquet ne doit pas être divisé pour faire une dose plus petite.
Prise de PROTONIX 20mg pour suspension buvable à la compote de pommes :
Prise de PROTONIX 40mg pour suspension buvable au jus de pomme :
Administration de PROTONIX 40 mg pour suspension buvable par sonde nasogastrique (NG) ou gastrostomie :
- PROTONIX pour suspension buvable peut être administré par une sonde nasogastrique ou une sonde de gastrostomie taille 16 français ou plus. Ne pas administrer PROTONIX 20 mg pour suspension buvable via une sonde NG ou une sonde de gastrostomie inférieure à la taille 16 French.
- Mélanger PROTONIX 20mg pour suspension buvable seulement dans le jus de pomme lors de l'administration via une sonde NG ou une sonde de gastrostomie.
Comment dois-je conserver PROTONIX 20 mg ?
Conservez PROTONIX 20mg à température ambiante entre 68°F et 77°F (20°C à 25°C).
Gardez PROTONIX 40 mg et tous les médicaments hors de la portée des enfants.
Ce guide de médication et ce mode d'emploi ont été approuvés par la Food and Drug Administration des États-Unis.