Xeloda 500mg Capecitabine Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce que Xeloda 500 mg et comment est-il utilisé ?
Xeloda est un médicament délivré sur ordonnance utilisé pour traiter les symptômes de cancers tels que le cancer du côlon, le cancer colorectal et le cancer du sein. Xeloda peut être utilisé seul ou avec d'autres médicaments.
Xeloda appartient à une classe de médicaments appelés antinéoplasiques, antimétabolites.
On ne sait pas si Xeloda 500 mg est sûr et efficace chez les enfants.
Quels sont les effets secondaires possibles de Xeloda 500 mg ?
Xeloda peut provoquer des effets secondaires graves, notamment :
- fièvre supérieure à 100,5 degrés,
- nausée,
- perte d'appétit,
- manger beaucoup moins que d'habitude,
- vomissements (plus d'une fois en 24 heures),
- diarrhée sévère (plus de 4 fois par jour, ou pendant la nuit),
- ampoules ou ulcères dans la bouche,
- gencives rouges ou enflées,
- difficulté à avaler,
- douleur, sensibilité, rougeur, gonflement, cloques ou desquamation de la peau des mains ou des pieds,
- avoir très soif ou chaud,
- être incapable d'uriner,
- transpiration abondante,
- peau chaude et sèche,
- douleur ou pression thoracique,
- battements cardiaques irréguliers,
- essoufflement,
- gonflement ou prise de poids rapide,
- miction douloureuse ou difficile,
- gonflement des pieds ou des chevilles,
- se sentir fatigué,
- essoufflement,
- urine foncée,
- selles de couleur argile,
- jaunissement de la peau ou des yeux (jaunisse),
- fièvre ou autres symptômes grippaux,
- toux,
- plaies cutanées,
- peau pâle,
- ecchymoses faciles,
- saignement inhabituel,
- se sentir étourdi,
- rythme cardiaque rapide,
- mal de gorge,
- gonflement du visage ou de la langue,
- brûlant dans tes yeux, et
- douleur cutanée suivie d'une éruption rouge ou violette (en particulier sur le visage ou le haut du corps) et provoquant des cloques et une desquamation
Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires les plus courants de Xeloda 500 mg incluent :
- Douleur d'estomac,
- constipation,
- maux d'estomac,
- sensation de fatigue,
- légère éruption cutanée, et
- engourdissement ou picotements dans les mains ou les pieds
Dites au médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles de Xeloda. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
ATTENTION
INTERACTION XELODA-WARFARINE
XELODA 500 mg warfarine Interaction : Les patients recevant de façon concomitante de la capécitabine et un traitement anticoagulant oral dérivé de la coumarine doivent faire surveiller fréquemment leur réponse anticoagulante (INR ou temps de prothrombine) afin d'ajuster la dose d'anticoagulant en conséquence. Une interaction médicamenteuse cliniquement importante entre XELODA et la warfarine a été démontrée dans un essai de pharmacologie clinique [voir AVERTISSEMENTS ET PRECAUTIONS et INTERACTIONS MÉDICAMENTEUSES]. Des paramètres de coagulation altérés et/ou des saignements, y compris la mort, ont été rapportés chez des patients prenant XELODA 500 mg en concomitance avec des anticoagulants dérivés de la coumarine tels que la warfarine et la phenprocoumone. Des rapports post-commercialisation ont montré des augmentations cliniquement significatives du temps de prothrombine (TP) et de l'INR chez des patients qui étaient stabilisés sous anticoagulants au moment de l'introduction de XELODA 500 mg. Ces événements sont survenus dans les jours et jusqu'à plusieurs mois après le début du traitement par XELODA 500 mg et, dans quelques cas, dans le mois suivant l'arrêt de XELODA. Ces événements sont survenus chez des patients avec et sans métastases hépatiques. Un âge supérieur à 60 ans et un diagnostic de cancer prédisposent indépendamment les patients à un risque accru de coagulopathie.
LA DESCRIPTION
XELODA (capécitabine) est un carbamate de fluoropyrimidine ayant une activité antinéoplasique. Il s'agit d'un promédicament systémique administré par voie orale de 5'-désoxy-5-fluorouridine (5'-DFUR) qui est converti en 5-fluorouracile.
Le nom chimique de la capécitabine est 5'-désoxy-5-fluoro-N-[(pentyloxy) carbonyl]-cytidine et a un poids moléculaire de 359,35. La capécitabine a la formule structurelle suivante :
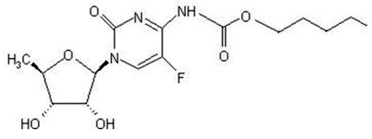
La capécitabine est une poudre cristalline blanche à blanc cassé avec une solubilité aqueuse de 26 mg/mL à 20 °C.
XELODA se présente sous forme de comprimés pelliculés biconvexes et oblongs pour administration orale. Chaque comprimé de couleur pêche clair contient 150 mg de capécitabine et chaque comprimé de couleur pêche contient 500 mg de capécitabine. Les ingrédients inactifs de XELODA comprennent : lactose anhydre, croscarmellose sodique, hydroxypropylméthylcellulose, cellulose microcristalline, stéarate de magnésium et eau purifiée. Le pelliculage pêche ou pêche clair contient de l'hydroxypropylméthylcellulose, du talc, du dioxyde de titane et des oxydes de fer jaune et rouge synthétiques.
LES INDICATIONS
Cancer colorectal
- XELODA est indiqué en tant qu'agent unique pour le traitement adjuvant chez les patients atteints d'un cancer du côlon de Dukes C ayant subi une résection complète de la tumeur primaire lorsqu'un traitement par fluoropyrimidine seule est préféré. XELODA 500 mg était non inférieur au 5fluorouracile et à la leucovorine (5-FU/LV) pour la survie sans maladie (DFS). Les médecins doivent tenir compte des résultats des essais de chimiothérapie combinée, qui ont montré une amélioration de la SSM et de la SG, lorsqu'ils prescrivent XELODA 500 mg en monothérapie dans le traitement adjuvant du cancer du côlon de Dukes C.
- XELODA 500 mg est indiqué comme traitement de première intention des patients atteints d'un carcinome colorectal métastatique lorsqu'un traitement par fluoropyrimidine seule est préféré. La chimiothérapie combinée a montré un bénéfice en termes de survie par rapport au 5-FU/LV seul. Un bénéfice en termes de survie par rapport au 5-FU/LV n'a pas été démontré avec XELODA 500 mg en monothérapie. L'utilisation de XELODA au lieu de 5FU/LV en association n'a pas été suffisamment étudiée pour assurer la sécurité ou la préservation de l'avantage de survie.
Cancer du sein
- XELODA en association au docétaxel est indiqué dans le traitement des patientes atteintes d'un cancer du sein métastatique après échec d'une chimiothérapie antérieure contenant des anthracyclines.
- XELODA 500 mg en monothérapie est également indiqué pour le traitement des patientes atteintes d'un cancer du sein métastatique résistant à la fois au paclitaxel et à une chimiothérapie contenant des anthracyclines ou résistantes au paclitaxel et pour lesquelles un traitement ultérieur par anthracyclines n'est pas indiqué (par exemple, les patientes ayant reçu des doses cumulées de 400 mg/m2 de doxorubicine ou d'équivalents de doxorubicine). La résistance est définie comme une maladie évolutive pendant le traitement, avec ou sans réponse initiale, ou une rechute dans les 6 mois suivant la fin du traitement avec un traitement adjuvant contenant de l'anthracycline.
DOSAGE ET ADMINISTRATION
Instructions d'administration importantes
Les comprimés XELODA doivent être avalés entiers avec de l'eau dans les 30 minutes qui suivent un repas. XELODA 500 mg est un médicament cytotoxique. Suivez les procédures spéciales de manipulation et d'élimination applicables.1 Si les comprimés de XELODA 500 mg doivent être coupés ou écrasés, cela doit être fait par un professionnel formé à la manipulation sûre des médicaments cytotoxiques en utilisant un équipement et des procédures de sécurité appropriés. La dose de XELODA est calculée en fonction de la surface corporelle.
Dose initiale standard
Monothérapie (cancer colorectal métastatique, cancer colorectal adjuvant, cancer du sein métastatique)
La dose recommandée de XELODA est de 1250 mg/m2 administrée par voie orale deux fois par jour (matin et soir ; équivalant à une dose quotidienne totale de 2500 mg/m2) pendant 2 semaines suivie d'une période de repos d'une semaine administrée en cycles de 3 semaines (voir ).
Le traitement adjuvant chez les patients atteints d'un cancer du côlon de Dukes' C est recommandé pour un total de 6 mois [c. de 8 cycles (24 semaines)].
En association avec le docétaxel (cancer du sein métastatique)
En association avec le docétaxel, la dose recommandée de XELODA est de 1250 mg/m2 deux fois par jour pendant 2 semaines suivie d'une période de repos d'une semaine, en association avec le docétaxel à 75 mg/m2 en perfusion intraveineuse d'une heure toutes les 3 semaines. Selon la notice du docétaxel, la prémédication doit être débutée avant l'administration du docétaxel chez les patients recevant l'association XELODA plus docétaxel. affiche la dose quotidienne totale de XELODA par surface corporelle et le nombre de comprimés à prendre à chaque prise.
Lignes directrices sur la gestion des doses
Général
La posologie de XELODA peut devoir être individualisée pour optimiser la prise en charge du patient. Les patients doivent être étroitement surveillés pour la toxicité et les doses de XELODA 500 mg doivent être modifiées si nécessaire pour s'adapter à la tolérance individuelle du patient au traitement [voir Etudes cliniques ]. La toxicité due à l'administration de XELODA 500 mg peut être gérée par un traitement symptomatique, des interruptions de dose et un ajustement de la dose de XELODA. Une fois la dose réduite, elle ne doit pas être augmentée ultérieurement. Les doses de XELODA 500 mg omises pour cause de toxicité ne sont ni remplacées ni rétablies ; au lieu de cela, le patient doit reprendre les cycles de traitement prévus.
La dose de phénytoïne et la dose d'anticoagulants dérivés de la coumarine peuvent devoir être réduites lorsque l'un ou l'autre médicament est administré en même temps que XELODA [voir INTERACTIONS MÉDICAMENTEUSES ].
Monothérapie (cancer colorectal métastatique, cancer colorectal adjuvant, cancer du sein métastatique)
Le schéma de modification de dose de XELODA tel que décrit ci-dessous (voir ) est recommandé pour la prise en charge des effets indésirables.
En association avec le docétaxel (cancer du sein métastatique)
Les modifications de dose de XELODA 500 mg pour cause de toxicité doivent être effectuées conformément à ce qui précède pour XELODA. Au début d'un cycle de traitement, si un délai de traitement est indiqué pour XELODA 500 mg ou le docétaxel, l'administration des deux agents doit être retardée jusqu'à ce que les conditions de redémarrage des deux médicaments soient remplies.
Le schéma de réduction de dose du docétaxel lorsqu'il est utilisé en association avec XELODA pour le traitement du cancer du sein métastatique est présenté dans le tableau 3.
Ajustement de la dose initiale dans les populations particulières
Insuffisance rénale
Aucun ajustement de la dose initiale de XELODA 500 mg n'est recommandé chez les patients présentant une insuffisance rénale légère (clairance de la créatinine = 51 à 80 mL/min [Cockroft et Gault, comme indiqué ci-dessous]). Chez les patients présentant une insuffisance rénale modérée (clairance initiale de la créatinine = 30 à 50 mL/min), une réduction de dose à 75 % de la dose initiale de XELODA lorsqu'il est utilisé en monothérapie ou en association avec le docétaxel (de 1250 mg/m2 à 950 mg/m2 deux fois par jour) est recommandé [voir Utilisation dans des populations spécifiques et PHARMACOLOGIE CLINIQUE ]. Un ajustement ultérieur de la dose est recommandé comme indiqué dans et (selon le régime) si un patient développe un événement indésirable de grade 2 à 4 [voir AVERTISSEMENTS ET PRECAUTIONS ]. Les recommandations d'ajustement de la dose initiale pour les patients présentant une insuffisance rénale modérée s'appliquent à la fois à XELODA 500 mg en monothérapie et à XELODA 500 mg en association avec le docétaxel.
Équation de Cockroft et Gault :
Gériatrie
Les médecins doivent faire preuve de prudence lors de la surveillance des effets de XELODA 500 mg chez les personnes âgées. Les données disponibles sont insuffisantes pour fournir une recommandation posologique.
COMMENT FOURNIE
Formes posologiques Points forts
XELODA se présente sous forme de comprimés pelliculés biconvexes et oblongs pour administration orale. Chaque comprimé de couleur pêche clair contient 150 mg de capécitabine et chaque comprimé de couleur pêche contient 500 mg de capécitabine.
150mg
Couleur : Pêche clair Gravure : XELODA d'un côté et 150 de l'autre Les comprimés de 150 mg sont conditionnés en flacons de 60 ( CDN 0004-1100-20), emballés individuellement dans un carton.
500mg
Couleur : Pêche Gravure : XELODA 500 mg d'un côté et 500 de l'autre Les comprimés de 500 mg sont conditionnés en flacons de 120 ( CDN 0004-1101-50), emballé individuellement dans un carton.
Stockage et manutention
Conserver à 25 °C (77 °F); excursions autorisées jusqu'à 15° à 30°C (59° à 86°F). [Voir USP Température ambiante contrôlée]. GARDER HERMÉTIQUEMENT FERMÉ.
XELODA est un médicament cytotoxique. Suivez les procédures spéciales applicables de manipulation et d'élimination.1 Tout produit non utilisé doit être éliminé conformément aux exigences locales ou aux programmes de reprise des médicaments.
RÉFÉRENCES
1. "Drogues dangereuses OSHA." OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
Distribué par : Genentech USA, Inc. Membre du groupe Roche, 1 DNA Way, South San Francisco, CA 94080-4990. Révisé : mai 2021
EFFETS SECONDAIRES
Étant donné que les essais cliniques sont menés dans des conditions très variables, les taux d'effets indésirables observés dans les essais cliniques d'un médicament ne peuvent pas être directement comparés aux taux des essais cliniques d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique.
Cancer du côlon adjuvant
montre les effets indésirables survenus chez ≥ 5 % des patients d'un essai de phase 3 chez des patients atteints d'un cancer du côlon de Dukes C qui ont reçu au moins une dose du médicament à l'étude et ont subi au moins une évaluation de l'innocuité. Au total, 995 patients ont été traités avec 1250 mg/m2 deux fois par jour de XELODA 500 mg administré pendant 2 semaines suivi d'une période de repos d'une semaine, et 974 patients ont reçu du 5-FU et de la leucovorine (20 mg/m2 de leucovorine IV suivi de 425 mg/m2 IV bolus 5FU les jours 1 à 5 tous les 28 jours). La durée médiane de traitement a été de 164 jours pour les patients traités par capécitabine et de 145 jours pour les patients traités par 5-FU/LV. Au total, 112 (11 %) et 73 (7 %) patients traités respectivement par la capécitabine et le 5-FU/LV ont arrêté le traitement en raison d'effets indésirables. Au total, 18 décès toutes causes confondues sont survenus pendant l'étude ou dans les 28 jours suivant l'administration du médicament à l'étude : 8 (0,8 %) patients randomisés pour XELODA 500 mg et 10 (1,0 %) randomisés pour 5-FU/LV.
montre des anomalies de laboratoire de grade 3/4 survenant chez ≥ 1 % des patients d'un essai de phase 3 chez des patients atteints d'un cancer du côlon de Dukes C qui ont reçu au moins une dose du médicament à l'étude et ont subi au moins une évaluation de l'innocuité.
Cancer colorectal métastatique
Monothérapie
montre les effets indésirables survenus chez ≥ 5 % des patients issus du regroupement des deux essais de phase 3 dans le cancer colorectal métastatique de première intention. Au total, 596 patients atteints d'un cancer colorectal métastatique ont été traités avec 1250 mg/m2 deux fois par jour de XELODA administré pendant 2 semaines suivi d'une période de repos d'une semaine, et 593 patients ont reçu du 5-FU et de la leucovorine dans le schéma Mayo (20 mg/m2 de leucovorine IV suivi d'un bolus IV de 425 mg/m2 de 5-FU, les jours 1 à 5, tous les 28 jours). Dans la base de données colorectale groupée, la durée médiane de traitement était de 139 jours pour les patients traités par capécitabine et de 140 jours pour les patients traités par 5-FU/LV. Au total, 78 (13 %) et 63 (11 %) patients traités par capécitabine et 5-FU/LV, respectivement, ont arrêté le traitement en raison d'effets indésirables/maladie intercurrente. Au total, 82 décès toutes causes confondues sont survenus pendant l'étude ou dans les 28 jours suivant l'administration du médicament à l'étude : 50 (8,4 %) patients randomisés pour XELODA 500 mg et 32 (5,4 %) randomisés pour 5-FU/LV.
Cancer du sein
En association avec le docétaxel
Les données suivantes sont présentées pour l'étude d'association avec XELODA 500 mg et le docétaxel chez des patientes atteintes d'un cancer du sein métastatique en et en . Dans le bras de l'association XELODA et docétaxel, le traitement était XELODA administré par voie orale à raison de 1250 mg/m2 deux fois par jour en traitement intermittent (2 semaines de traitement suivies d'une semaine sans traitement) pendant au moins 6 semaines et le docétaxel administré en perfusion intraveineuse d'une heure à une dose de 75 mg/m2 le premier jour de chaque cycle de 3 semaines pendant au moins 6 semaines. Dans le bras monothérapie, le docétaxel a été administré en perfusion intraveineuse d'une heure à une dose de 100 mg/m2 le premier jour de chaque cycle de 3 semaines pendant au moins 6 semaines. La durée moyenne de traitement a été de 129 jours dans le bras association et de 98 jours dans le bras monothérapie. Au total, 66 patients (26 %) dans le bras association et 49 (19 %) dans le bras monothérapie se sont retirés de l'étude en raison d'effets indésirables. Le pourcentage de patients nécessitant des réductions de dose en raison d'effets indésirables était de 65 % dans le bras association et de 36 % dans le bras monothérapie. Le pourcentage de patients nécessitant des interruptions de traitement en raison d'effets indésirables dans le bras combiné était de 79 %. Les interruptions de traitement faisaient partie du schéma de modification de dose pour le bras de thérapie combinée mais pas pour les patients traités par le docétaxel en monothérapie.
Monothérapie
Les données suivantes sont présentées pour l'étude chez des patientes atteintes d'un cancer du sein de stade IV qui ont reçu une dose de 1250 mg/m2 administrée deux fois par jour pendant 2 semaines, suivie d'une période de repos d'une semaine. La durée moyenne du traitement était de 114 jours. Au total, 13 patients sur 162 (8 %) ont interrompu le traitement en raison d'effets indésirables/maladies intercurrentes.
Événements indésirables cliniquement pertinents chez
Les événements indésirables cliniquement pertinents signalés chez
Monothérapie (cancer colorectal métastatique, cancer colorectal adjuvant, cancer du sein métastatique)
Gastro-intestinal : distension abdominale, dysphagie, proctalgie, ascite (0,1 %), ulcère gastrique (0,1 %), iléus (0,3 %), dilatation toxique de l'intestin, gastro-entérite (0,1 %)
Peau et sous-cutanée : trouble des ongles (0,1 %), augmentation de la transpiration (0,1 %), réaction de photosensibilité (0,1 %), ulcération cutanée, prurit, syndrome de rappel radiologique (0,2 %)
Général: douleur thoracique (0,2 %), syndrome grippal, bouffées de chaleur, douleur (0,1 %), enrouement, irritabilité, difficulté à marcher, soif, masse thoracique, collapsus, fibrose (0,1 %), hémorragie, œdème, sédation
Neurologique : insomnie, ataxie (0,5 %), tremblements, dysphasie, encéphalopathie (0,1 %), troubles de la coordination, dysarthrie, perte de conscience (0,2 %), troubles de l'équilibre
Métabolisme: prise de poids, cachexie (0,4 %), hypertriglycéridémie (0,1 %), hypokaliémie, hypomagnésémie
Œil: conjonctivite
Respiratoire: toux (0,1 %), épistaxis (0,1 %), asthme (0,2 %), hémoptysie, détresse respiratoire (0,1 %), dyspnée
Cardiaque: tachycardie (0,1 %), bradycardie, fibrillation auriculaire, extrasystoles ventriculaires, extrasystoles, myocardite (0,1 %), épanchement péricardique
Infections : laryngite (1,0 %), bronchite (0,2 %), pneumonie (0,2 %), bronchopneumonie (0,2 %), kératoconjonctivite, septicémie (0,3 %), infections fongiques (dont candidose) (0,2 %)
Musculo-squelettique : myalgies, douleurs osseuses (0,1 %), arthrite (0,1 %), faiblesse musculaire
Sang & Lymphatique : leucopénie (0,2 %), trouble de la coagulation (0,1 %), aplasie médullaire (0,1 %), purpura thrombocytopénique idiopathique (1,0 %), pancytopénie (0,1 %)
Vasculaire: hypotension (0,2 %), hypertension (0,1 %), lymphœdème (0,1 %), embolie pulmonaire (0,2 %), accident vasculaire cérébral (0,1 %)
Psychiatrique: dépression, confusion (0,1 %)
Rénal: insuffisance rénale (0,6 %)
Oreille: vertige
Hépatobiliaire : fibrose hépatique (0,1 %), hépatite (0,1 %), hépatite cholestatique (0,1 %), anomalies des tests de la fonction hépatique
Système immunitaire: hypersensibilité médicamenteuse (0,1 %)
XELODA 500 mg en association avec le docétaxel (cancer du sein métastatique)
Gastro-intestinal : iléus (0,4 %), entérocolite nécrosante (0,4 %), ulcère de l'œsophage (0,4 %), diarrhée hémorragique (0,8 %)
Neurologique : ataxie (0,4 %), syncope (1,2 %), perte du goût (0,8 %), polyneuropathie (0,4 %), migraine (0,4 %)
Cardiaque: tachycardie supraventriculaire (0,4 %)
Infection: sepsis neutropénique (2,4 %), sepsis (0,4 %), bronchopneumonie (0,4 %)
Lymphatique: agranulocytose (0,4 %), diminution de la prothrombine (0,4 %)
Vasculaire: hypotension (1,2 %), phlébite et thrombophlébite veineuses (0,4 %), hypotension orthostatique (0,8 %)
Rénal: insuffisance rénale (0,4%)
Hépatobiliaire : ictère (0,4 %), anomalies des tests de la fonction hépatique (0,4 %), insuffisance hépatique (0,4 %), coma hépatique (0,4 %), hépatotoxicité (0,4 %)
Système immunitaire: hypersensibilité (1,2%)
Expérience post-commercialisation
Les effets indésirables suivants ont été observés depuis la commercialisation : œdème de Quincke, insuffisance hépatique, sténose du canal lacrymal, insuffisance rénale aiguë secondaire à une déshydratation, y compris fatale [voir AVERTISSEMENTS ET PRECAUTIONS [voir AVERTISSEMENTS ET PRECAUTIONS ], le syndrome main-pied persistant ou grave peut éventuellement entraîner la perte des empreintes digitales [voir AVERTISSEMENTS ET PRECAUTIONS ]
En cas d'exposition à des comprimés de XELODA 500 mg écrasés, les effets indésirables suivants ont été signalés : irritation et gonflement des yeux, éruption cutanée, diarrhée, paresthésie, maux de tête, irritation gastrique, vomissements et nausées.
INTERACTIONS MÉDICAMENTEUSES
Interactions médicament-médicament
Anticoagulants
Des paramètres de coagulation altérés et/ou des saignements ont été rapportés chez des patients prenant XELODA 500 mg en concomitance avec des anticoagulants dérivés de la coumarine tels que la warfarine et la phenprocoumone [voir AVERTISSEMENT DE BOÎTE ]. Ces événements sont survenus dans les jours et jusqu'à plusieurs mois après le début du traitement par XELODA 500 mg et, dans quelques cas, dans le mois suivant l'arrêt de XELODA. Ces événements sont survenus chez des patients avec et sans métastases hépatiques. Dans une étude d'interaction médicamenteuse avec l'administration d'une dose unique de warfarine, il y a eu une augmentation significative de l'ASC moyenne de la S-warfarine [voir PHARMACOLOGIE CLINIQUE ]. La valeur maximale de l'INR observée a augmenté de 91 %. Cette interaction est probablement due à une inhibition du cytochrome P450 2C9 par la capécitabine et/ou ses métabolites.
Phénytoïne
Le taux de phénytoïne doit être étroitement surveillé chez les patients prenant XELODA 500 mg et il peut être nécessaire de réduire la dose de phénytoïne [voir DOSAGE ET ADMINISTRATION ]. Des rapports post-commercialisation indiquent que certains patients recevant XELODA 500 mg et de la phénytoïne ont présenté une toxicité associée à des taux élevés de phénytoïne. Aucune étude formelle d'interaction médicamenteuse avec la phénytoïne n'a été menée, mais le mécanisme d'interaction est présumé être l'inhibition de l'isoenzyme CYP2C9 par la capécitabine et/ou ses métabolites.
Leucovorine
La concentration de 5-fluorouracile est augmentée et sa toxicité peut être augmentée par la leucovorine. Des décès par entérocolite grave, diarrhée et déshydratation ont été signalés chez des patients âgés recevant de la leucovorine et du fluorouracile une fois par semaine.
Substrats du CYP2C9
Hormis la warfarine, aucune étude formelle sur les interactions médicamenteuses entre XELODA et d'autres substrats du CYP2C9 n'a été menée. La prudence s'impose lorsque XELODA est co-administré avec des substrats du CYP2C9.
Allopurinol
L'utilisation concomitante avec l'allopurinol peut diminuer la concentration des métabolites actifs de la capécitabine [voir PHARMACOLOGIE CLINIQUE ], ce qui peut diminuer l'efficacité de XELODA. Éviter l'utilisation d'allopurinol pendant le traitement par XELODA.
Interaction médicament-aliment
Il a été démontré que la nourriture réduisait à la fois le taux et l'étendue de l'absorption de la capécitabine [voir PHARMACOLOGIE CLINIQUE ]. Dans tous les essais cliniques, les patients devaient administrer XELODA 500 mg dans les 30 minutes suivant un repas. Il est recommandé d'administrer XELODA avec de la nourriture [voir DOSAGE ET ADMINISTRATION ].
AVERTISSEMENTS
Inclus dans le cadre du "PRÉCAUTIONS" Section
PRÉCAUTIONS
Coagulopathie
Les patients recevant simultanément de la capécitabine et un traitement anticoagulant dérivé de la coumarine par voie orale doivent faire surveiller étroitement leur réponse anticoagulante (INR ou temps de prothrombine) et la dose d'anticoagulant doit être ajustée en conséquence [voir AVERTISSEMENT DE BOÎTE et INTERACTIONS MÉDICAMENTEUSES ].
Diarrhée
XELODA 500mg peut induire des diarrhées, parfois sévères. Les patients souffrant de diarrhée sévère doivent être étroitement surveillés et recevoir un remplacement hydrique et électrolytique s'ils se déshydratent. Chez 875 patients atteints d'un cancer du sein métastatique ou d'un cancer colorectal ayant reçu XELODA en monothérapie, le délai médian avant la première apparition d'une diarrhée de grade 2 à 4 était de 34 jours (intervalle de 1 à 369 jours). La durée médiane des diarrhées de grade 3 à 4 était de 5 jours. La diarrhée de grade 2 de l'Institut national du cancer du Canada (INCC) est définie comme une augmentation de 4 à 6 selles/jour ou de selles nocturnes, la diarrhée de grade 3 comme une augmentation de 7 à 9 selles/jour ou l'incontinence et la malabsorption, et la diarrhée de grade 4 comme une augmentation de ≥ 10 selles/jour ou une diarrhée sanglante ou la nécessité d'une assistance parentérale. En cas de diarrhée de grade 2, 3 ou 4, l'administration de XELODA doit être immédiatement interrompue jusqu'à ce que la diarrhée disparaisse ou diminue d'intensité jusqu'au grade 1 [voir DOSAGE ET ADMINISTRATION ]. Les traitements antidiarrhéiques standards (p. ex., lopéramide) sont recommandés.
Une entérocolite nécrosante (typhlite) a été rapportée.
Cardiotoxicité
La cardiotoxicité observée avec XELODA 500 mg comprend l'infarctus du myocarde/l'ischémie, l'angor, les troubles du rythme, l'arrêt cardiaque, l'insuffisance cardiaque, la mort subite, les modifications électrocardiographiques et la cardiomyopathie. Ces effets indésirables peuvent être plus fréquents chez les patients ayant des antécédents de maladie coronarienne.
Déficit en dihydropyrimidine déshydrogénase
D'après les rapports post-commercialisation, les patients présentant certaines mutations homozygotes ou certaines mutations hétérozygotes composées du gène DPD qui entraînent une absence complète ou presque complète de l'activité DPD présentent un risque accru de toxicité aiguë précoce et d'effets indésirables graves, potentiellement mortels ou mortels. réactions causées par XELODA (p. ex. mucosite, diarrhée, neutropénie et neurotoxicité). Les patients présentant une activité DPD partielle peuvent également présenter un risque accru d'effets indésirables graves, potentiellement mortels ou mortels causés par XELODA.
Suspendre ou arrêter définitivement XELODA 500 mg sur la base d'une évaluation clinique de l'apparition, de la durée et de la sévérité des toxicités observées chez les patients présentant des signes de toxicité aiguë précoce ou inhabituellement sévère, pouvant indiquer une absence quasi complète ou totale d'activité DPD. Aucune dose de XELODA n'a fait la preuve de son innocuité chez les patients en absence totale d'activité DPD. Il n'y a pas suffisamment de données pour recommander une dose spécifique chez les patients présentant une activité DPD partielle telle que mesurée par un test spécifique.
Déshydratation et insuffisance rénale
Une déshydratation a été observée et peut provoquer une insuffisance rénale aiguë qui peut être mortelle. Les patients présentant une fonction rénale altérée préexistante ou qui reçoivent XELODA en concomitance avec des agents néphrotoxiques connus sont plus à risque. Les patients souffrant d'anorexie, d'asthénie, de nausées, de vomissements ou de diarrhée peuvent rapidement se déshydrater. Surveiller les patients lors de l'administration de XELODA 500 mg pour prévenir et corriger la déshydratation au début. En cas de déshydratation de grade 2 (ou supérieur), le traitement par XELODA doit être immédiatement interrompu et la déshydratation corrigée. Le traitement ne doit pas être repris tant que le patient n'est pas réhydraté et que toutes les causes déclenchantes n'ont pas été corrigées ou contrôlées. Des modifications de dose doivent être appliquées pour l'événement indésirable précipitant si nécessaire [voir DOSAGE ET ADMINISTRATION ].
Les patients présentant une insuffisance rénale modérée au départ nécessitent une réduction de la dose [voir DOSAGE ET ADMINISTRATION ]. Les patients présentant une insuffisance rénale légère à modérée au départ doivent être étroitement surveillés afin de détecter tout effet indésirable. Une interruption rapide du traitement suivie d'ajustements posologiques ultérieurs est recommandée si un patient développe un événement indésirable de grade 2 à 4, comme indiqué dans [voir DOSAGE ET ADMINISTRATION , Utilisation dans des populations spécifiques , et PHARMACOLOGIE CLINIQUE ].
Toxicité embryo-fœtale
D'après les résultats d'études sur la reproduction animale et son mécanisme d'action, XELODA peut nuire au fœtus lorsqu'il est administré à une femme enceinte [voir PHARMACOLOGIE CLINIQUE ]. Les données limitées disponibles ne sont pas suffisantes pour informer l'utilisation de XELODA 500 mg chez la femme enceinte. Dans les études de reproduction chez l'animal, l'administration de capécitabine à des femelles gravides pendant la période d'organogenèse a entraîné une embryolétalité et une tératogénicité chez la souris et une embryolétalité chez le singe à 0,2 et 0,6 fois l'exposition (AUC) chez les patients recevant respectivement la dose recommandée [voir Utilisation dans des populations spécifiques ]. Informer les femmes enceintes du risque potentiel pour le fœtus. Conseiller aux femmes en âge de procréer d'utiliser une contraception efficace pendant le traitement et pendant les 6 mois suivant la dernière dose de XELODA [voir Utilisation dans des populations spécifiques ].
Toxicité mucocutanée et dermatologique
Des réactions cutanéo-muqueuses sévères, dont certaines avec une issue fatale, telles que le syndrome de Stevens-Johnson et la nécrolyse épidermique toxique (NET) peuvent survenir chez les patients traités par XELODA [voir EFFETS INDÉSIRABLES ]. XELODA 500 mg doit être arrêté définitivement chez les patients qui présentent une réaction cutanéo-muqueuse sévère pouvant être attribuée au traitement par XELODA.
Le syndrome mains-pieds (érythrodysesthésie palmo-plantaire ou érythème acral induit par la chimiothérapie) est une toxicité cutanée. Le délai médian d'apparition était de 79 jours (intervalle de 11 à 360 jours) avec une échelle de gravité de grades 1 à 3 pour les patients recevant XELODA 500 mg en monothérapie dans un contexte métastatique. Le grade 1 est caractérisé par l'un des éléments suivants : engourdissement, dysesthésie/paresthésie, picotements, gonflement indolore ou érythème des mains et/ou des pieds et/ou inconfort qui ne perturbe pas les activités normales. Le syndrome mains-pieds de grade 2 est défini comme un érythème douloureux et un gonflement des mains et/ou des pieds et/ou une gêne affectant les activités de la vie quotidienne du patient. Le syndrome mains-pieds de grade 3 est défini comme une desquamation humide, une ulcération, des cloques ou une douleur intense des mains et/ou des pieds et/ou un inconfort sévère qui empêche le patient de travailler ou d'accomplir les activités de la vie quotidienne. Le syndrome mains-pieds persistant ou sévère (grade 2 et plus) peut éventuellement entraîner la perte des empreintes digitales, ce qui pourrait avoir un impact sur l'identification du patient. En cas de survenue d'un syndrome main-pied de grade 2 ou 3, l'administration de XELODA doit être interrompue jusqu'à ce que l'événement disparaisse ou diminue d'intensité au grade 1. Après un syndrome main-pied de grade 3, les doses suivantes de XELODA 500 mg doivent être diminuées [ voir DOSAGE ET ADMINISTRATION ].
Hyperbilirubinémie
Chez 875 patients atteints d'un cancer du sein métastatique ou d'un cancer colorectal ayant reçu au moins une dose de XELODA 1250 mg/m2 deux fois par jour en monothérapie pendant 2 semaines suivies d'une période de repos d'une semaine, une hyperbilirubinémie de grade 3 (1,5-3 x LSN) est survenue chez 15,2 % (n = 133) des patients et une hyperbilirubinémie de grade 4 (> 3 x LSN) sont survenues chez 3,9 % (n = 34) des patients. Sur 566 patients qui avaient des métastases hépatiques au départ et 309 patients sans métastases hépatiques au départ, une hyperbilirubinémie de grade 3 ou 4 est survenue chez 22,8 % et 12,3 %, respectivement. Parmi les 167 patients atteints d'hyperbilirubinémie de grade 3 ou 4, 18,6 % (n = 31) présentaient également des élévations post-inclusion (grades 1 à 4, sans élévation au départ) de la phosphatase alcaline et 27,5 % (n = 46) présentaient des élévations post-inclusion des transaminases au à tout moment (pas nécessairement simultanément). La majorité de ces patients, 64,5 % (n = 20) et 71,7 % (n = 33), avaient des métastases hépatiques au départ. En outre, 57,5 % (n = 96) et 35,3 % (n = 59) des 167 patients présentaient des élévations (grades 1 à 4) à la fois avant et après l'inclusion des phosphatases alcalines ou des transaminases, respectivement. Seuls 7,8 % (n = 13) et 3,0 % (n = 5) avaient des élévations de grade 3 ou 4 de la phosphatase alcaline ou des transaminases.
Chez les 596 patients traités par XELODA 500 mg en traitement de première intention du cancer colorectal métastatique, l'incidence de l'hyperbilirubinémie de grade 3 ou 4 était similaire à la base de données globale sur l'innocuité des essais cliniques de XELODA 500 mg en monothérapie. Le délai médian d'apparition de l'hyperbilirubinémie de grade 3 ou 4 dans la population atteinte de cancer colorectal était de 64 jours et la bilirubine totale médiane est passée de 8 μm/L au départ à 13 μm/L pendant le traitement par XELODA. Sur les 136 patients atteints d'un cancer colorectal présentant une hyperbilirubinémie de grade 3 ou 4, 49 patients avaient une hyperbilirubinémie de grade 3 ou 4 comme dernière valeur mesurée, dont 46 avaient des métastases hépatiques au départ.
Chez 251 patientes atteintes d'un cancer du sein métastatique ayant reçu une association de XELODA 500 mg et de docétaxel, une hyperbilirubinémie de grade 3 (1,5 à 3 x LSN) est survenue chez 7 % (n = 17) et une hyperbilirubinémie de grade 4 (> 3 x LSN) est survenue chez 2 % (n=5).
En cas d'élévations de la bilirubine de grade 3 à 4 liées au médicament, l'administration de XELODA 500 mg doit être immédiatement interrompue jusqu'à ce que l'hyperbilirubinémie diminue à ≤ 3,0 X LSN [voir les modifications posologiques recommandées sous DOSAGE ET ADMINISTRATION ].
Hématologique
Chez 875 patients atteints d'un cancer du sein métastatique ou d'un cancer colorectal qui ont reçu une dose de 1 250 mg/m2 administrée deux fois par jour en monothérapie pendant 2 semaines suivies d'une période de repos d'une semaine, 3,2 %, 1,7 % et 2,4 % des patients avaient un grade 3 ou 4 neutropénie, thrombocytopénie ou diminution de l'hémoglobine, respectivement. Chez 251 patientes atteintes d'un cancer du sein métastatique ayant reçu une dose de XELODA en association avec le docétaxel, 68 % présentaient une neutropénie de grade 3 ou 4, 2,8 % présentaient une thrombocytopénie de grade 3 ou 4 et 9,6 % présentaient une anémie de grade 3 ou 4.
Les patients dont le nombre initial de neutrophiles est
Patients gériatriques
Les patients âgés de ≥ 80 ans peuvent présenter une plus grande incidence d'effets indésirables de grade 3 ou 4. Chez 875 patients atteints d'un cancer du sein métastatique ou d'un cancer colorectal ayant reçu XELODA 500 mg en monothérapie, 62 % des 21 patients ≥ 80 ans traités par XELODA 500 mg ont présenté un événement indésirable de grade 3 ou 4 lié au traitement : diarrhée chez 6 (28,6 %) , des nausées chez 3 (14,3 %), un syndrome main-pied chez 3 (14,3 %) et des vomissements chez 2 (9,5 %) patients. Parmi les 10 patients âgés de 70 ans et plus (aucun patient n'avait plus de 80 ans) traités par XELODA 500 mg en association avec le docétaxel, 30 % (3 sur 10) des patients ont présenté une diarrhée et une stomatite de grade 3 ou 4, et 40 % (4 sur 10) ont présenté un syndrome mains-pieds de grade 3.
Parmi les 67 patients âgés de ≥ 60 ans recevant XELODA en association avec le docétaxel, l'incidence des effets indésirables de grade 3 ou 4 liés au traitement, des effets indésirables graves liés au traitement, des abandons en raison d'effets indésirables, des arrêts de traitement en raison d'effets indésirables et du traitement les interruptions au cours des deux premiers cycles de traitement étaient plus élevées que dans le groupe de patients
Chez 995 patients recevant XELODA en traitement adjuvant du cancer du côlon de Dukes C après résection de la tumeur primitive, 41 % des 398 patients âgés de ≥ 65 ans traités par XELODA ont présenté un événement indésirable de grade 3 ou 4 lié au traitement : - syndrome du pied chez 75 (18,8 %), diarrhée chez 52 (13,1 %), stomatite chez 12 (3,0 %), neutropénie/granulocytopénie chez 11 (2,8 %), vomissements chez 6 (1,5 %) et nausées chez 5 (1,3 %) les patients. Chez les patients âgés de ≥ 65 ans (ensemble de la population randomisée ; capécitabine 188 patients, 5-FU/LV 208 patients) traités pour un cancer du côlon de Dukes C après résection de la tumeur primitive, les rapports de risque pour la survie sans maladie et la survie globale pour XELODA par rapport au 5-FU/LV étaient de 1,01 (IC à 95 % 0,80 – 1,27) et 1,04 (IC à 95 % 0,79 – 1,37), respectivement.
Insuffisance hépatique
Les patients présentant un dysfonctionnement hépatique léger à modéré dû à des métastases hépatiques doivent être étroitement surveillés lors de l'administration de XELODA. L'effet d'un dysfonctionnement hépatique sévère sur l'élimination de XELODA n'est pas connu [voir Utilisation dans des populations spécifiques et PHARMACOLOGIE CLINIQUE ].
Combinaison avec d'autres médicaments
L'utilisation de XELODA en association avec l'irinotécan n'a pas été suffisamment étudiée.
Informations sur les conseils aux patients
Conseillez au patient de lire l'étiquetage patient approuvé par la FDA ( INFORMATIONS PATIENTS ).
Diarrhée
Informez les patients souffrant de diarrhée de grade 2 (augmentation de 4 à 6 selles/jour ou selles nocturnes) ou plus ou souffrant de diarrhée sanglante sévère avec douleurs abdominales sévères et fièvre d'arrêter de prendre XELODA. Conseiller les patients sur l'utilisation de traitements antidiarrhéiques (par exemple, le lopéramide) pour gérer la diarrhée [voir AVERTISSEMENTS ET PRECAUTIONS ].
Cardiotoxicité
Avertir les patients du risque de cardiotoxicité et de contacter immédiatement leur fournisseur de soins de santé ou de se rendre aux urgences en cas de nouvelle apparition de douleurs thoraciques, d'essoufflement, d'étourdissements ou d'étourdissements [voir AVERTISSEMENTS ET PRECAUTIONS ].
Déficit en dihydropyrimidine déshydrogénase
Conseillez aux patients d'informer leur fournisseur de soins de santé s'ils ont un déficit connu en DPD. Aviser les patients s'ils présentent une absence complète ou quasi complète d'activité DPD qu'ils courent un risque accru de toxicité aiguë précoce et d'effets indésirables graves, potentiellement mortels ou mortels causés par XELODA (p. ex., mucosite, diarrhée, neutropénie et neurotoxicité) [voir AVERTISSEMENTS ET PRECAUTIONS ].
Déshydratation et insuffisance rénale
Demandez aux patients souffrant de déshydratation de grade 2 ou plus (liquides IV indiqués AVERTISSEMENTS ET PRECAUTIONS ].
Instructions d'administration importantes
Conseillez aux patients d'avaler les comprimés de XELODA 500 mg entiers avec de l'eau dans les 30 minutes qui suivent un repas. Conseillez aux patients et aux soignants de ne pas écraser ou couper les comprimés XELODA. S'ils ne peuvent pas avaler les comprimés de XELODA en entier, conseillez aux patients d'en informer leur professionnel de la santé [voir DOSAGE ET ADMINISTRATION ].
Hypersensibilité et œdème de Quincke
Informer les patients que XELODA peut provoquer des réactions d'hypersensibilité sévères et un œdème de Quincke. Conseillez aux patients qui ont une hypersensibilité connue à la capécitabine ou au 5-fluorouracile d'informer leur médecin [voir CONTRE-INDICATIONS ]. Demandez aux patients qui développent des réactions d'hypersensibilité ou des symptômes cutanéo-muqueux (p. ex., urticaire, éruption cutanée, érythème, prurit ou gonflement du visage, des lèvres, de la langue ou de la gorge rendant difficile la déglutition ou la respiration) d'arrêter de prendre XELODA et de contacter immédiatement leur professionnel de la santé ou aller aux urgences. [voir EFFETS INDÉSIRABLES ].
Nausée
Demandez aux patients souffrant de nausées de grade 2 (apport alimentaire significativement diminué mais capables de manger par intermittence) ou plus d'arrêter immédiatement de prendre XELODA 500 mg et de contacter leur professionnel de la santé pour la prise en charge des nausées [voir EFFETS INDÉSIRABLES ].

Vomissement
Demandez aux patients présentant des vomissements de grade 2 (2 à 5 épisodes sur une période de 24 heures) ou plus d'arrêter de prendre XELODA immédiatement et de contacter leur fournisseur de soins de santé pour la prise en charge des vomissements [voir EFFETS INDÉSIRABLES ].
Syndrome main-pied
Demandez aux patients souffrant d'un syndrome main-pied de grade 2 (érythème douloureux et gonflement des mains et/ou des pieds et/ou inconfort affectant les activités de la vie quotidienne du patient) ou plus d'arrêter immédiatement de prendre XELODA 500 mg et de contacter leur professionnel de la santé . Informez les patients que l'instauration d'un traitement symptomatique est recommandée, et que le syndrome mains-pieds peut entraîner la perte d'empreintes digitales, ce qui pourrait avoir un impact sur l'identification personnelle [voir EFFETS INDÉSIRABLES ].
Stomatite
Informez les patients souffrant de stomatite de grade 2 (érythème douloureux, œdème ou ulcères de la bouche ou de la langue, mais capables de manger) ou plus d'arrêter immédiatement de prendre XELODA et de contacter leur professionnel de la santé [voir EFFETS INDÉSIRABLES ].
fièvre et neutropénie
Informez les patients qui développent une fièvre de 100,5 °F ou plus ou tout autre signe d'infection potentielle de contacter leur fournisseur de soins de santé [voir EFFETS INDÉSIRABLES ].
Toxicité embryo-fœtale
Informer les femmes en âge de procréer du risque potentiel pour le fœtus et d'utiliser une contraception efficace pendant le traitement par XELODA 500 mg et pendant 6 mois après la dernière dose. Conseillez aux femmes d'informer leur fournisseur de soins de santé d'une grossesse connue ou suspectée [voir AVERTISSEMENTS ET PRECAUTIONS , Utilisation dans des populations spécifiques ].
Conseiller aux patients de sexe masculin ayant des partenaires féminines en âge de procréer d'utiliser une contraception efficace pendant le traitement par XELODA 500 mg et pendant les 3 mois suivant la dernière dose [voir Utilisation dans des populations spécifiques ].
Lactation
Déconseiller aux femmes d'allaiter pendant le traitement par XELODA 500 mg et pendant 2 semaines après la dernière dose [voir Utilisation dans des populations spécifiques ].
Toxicologie non clinique
Carcinogenèse, mutagenèse, altération de la fertilité
Des études adéquates portant sur le potentiel carcinogène de la capécitabine n'ont pas été menées. La capécitabine n'a pas été mutagène in vitro sur les bactéries (test d'Ames) ou les cellules de mammifères (test de mutation du gène V79/HPRT de hamster chinois). La capécitabine était clastogène in vitro sur les lymphocytes du sang périphérique humain mais pas clastogène in vivo sur la moelle osseuse de souris (test du micronoyau). Le fluorouracile provoque des mutations dans les bactéries et les levures. Le fluorouracile provoque également des anomalies chromosomiques dans le test du micronoyau de la souris in vivo.
Dans des études sur la fertilité et les performances générales de reproduction chez des souris femelles, des doses orales de capécitabine de 760 mg/kg/jour (environ 2300 mg/m2/jour) ont perturbé l'œstrus et ont par conséquent entraîné une diminution de la fertilité. Chez les souris qui sont devenues enceintes, aucun fœtus n'a survécu à cette dose. La perturbation de l'œstrus était réversible. Chez les mâles, cette dose a provoqué des modifications dégénératives des testicules, notamment une diminution du nombre de spermatocytes et de spermatides. Dans des études pharmacocinétiques distinctes, cette dose chez la souris a produit des valeurs d'ASC du 5'-DFUR d'environ 0,7 fois les valeurs correspondantes chez les patients ayant reçu la dose quotidienne recommandée.
Utilisation dans des populations spécifiques
Grossesse
Résumé des risques
D'après les résultats d'études sur la reproduction animale et son mécanisme d'action, XELODA 500 mg peut nuire au fœtus lorsqu'il est administré à une femme enceinte [voir PHARMACOLOGIE CLINIQUE ]. Les données humaines limitées disponibles ne sont pas suffisantes pour informer le risque associé au médicament pendant la grossesse. Dans les études de reproduction chez l'animal, l'administration de capécitabine à des femelles gravides pendant la période d'organogenèse a entraîné une létalité embryonnaire et une tératogénicité chez la souris et une létalité embryonnaire chez le singe à 0,2 et 0,6 fois l'exposition (AUC) chez les patients recevant la dose recommandée respectivement [voir Données ]. Informer les femmes enceintes du risque potentiel pour le fœtus.
Le risque de fond estimé de malformations congénitales majeures et de fausse couche pour la population indiquée est inconnu. Dans la population générale des États-Unis, le risque de fond estimé de malformations congénitales majeures et de fausse couche dans les grossesses cliniquement reconnues est de 2 à 4 % et de 15 à 20 %, respectivement.
Données
Données animales
L'administration orale de capécitabine à des souris gravides pendant la période d'organogenèse à la dose de 198 mg/kg/jour a entraîné des malformations et une létalité embryonnaire. Dans des études pharmacocinétiques distinctes, cette dose chez la souris a produit des valeurs d'ASC du 5'-DFUR qui étaient environ 0,2 fois les valeurs d'ASC chez les patients ayant reçu la dose quotidienne recommandée. Les malformations chez les souris comprenaient la fente palatine, l'anophtalmie, la microphtalmie, l'oligodactylie, la polydactylie, la syndactylie, la queue crépue et la dilatation des ventricules cérébraux. L'administration orale de capécitabine à des singes gravides pendant la période d'organogenèse à une dose de 90 mg/kg/jour a entraîné une létalité fœtale. Cette dose a produit des valeurs d'AUC de 5'-DFUR qui étaient environ 0,6 fois les valeurs d'AUC chez les patients ayant reçu la dose quotidienne recommandée.
Lactation
Résumé des risques
Il n'existe aucune information concernant la présence de capécitabine dans le lait maternel, ni sur ses effets sur la production de lait ou sur le nourrisson allaité. Des métabolites de la capécitabine étaient présents dans le lait de souris en lactation [voir Données ]. En raison du risque d'effets indésirables graves liés à l'exposition à la capécitabine chez les nourrissons allaités, déconseiller aux femmes d'allaiter pendant le traitement par XELODA 500 mg et pendant 2 semaines après la dernière dose.
Données
Des souris allaitantes ayant reçu une dose orale unique de capécitabine ont excrété des quantités significatives de métabolites de capécitabine dans le lait.
Femelles et mâles de potentiel reproducteur
Test de grossesse
Un test de grossesse est recommandé pour les femmes en âge de procréer avant de commencer XELODA.
La contraception
Femelles
XELODA 500 mg peut nuire au fœtus lorsqu'il est administré à une femme enceinte [voir Grossesse ]. Conseillez aux femmes en âge de procréer d'utiliser une contraception efficace pendant le traitement et pendant les 6 mois suivant la dernière dose de XELODA.
Mâles
Sur la base des résultats de toxicité génétique, conseiller aux patients masculins ayant des partenaires féminines en âge de procréer d'utiliser une contraception efficace pendant le traitement et pendant les 3 mois suivant la dernière dose de XELODA [voir Toxicologie non clinique ].
Infertilité
D'après des études sur des animaux, XELODA peut altérer la fertilité chez les femelles et les mâles en âge de procréer [voir Toxicologie non clinique ].
Utilisation pédiatrique
L'innocuité et l'efficacité de XELODA chez les patients pédiatriques n'ont pas été établies. Aucun bénéfice clinique n'a été démontré dans deux essais à un seul bras chez des patients pédiatriques atteints de gliomes du tronc cérébral nouvellement diagnostiqués et de gliomes de haut grade. Dans les deux essais, les patients pédiatriques ont reçu une formulation pédiatrique expérimentale de capécitabine en concomitance avec et après la fin de la radiothérapie (dose totale de 5580 cGy en fractions de 180 cGy). La biodisponibilité relative de la formulation expérimentale par rapport à XELODA 500 mg était similaire.
Le premier essai a été mené chez 22 patients pédiatriques (âge médian 8 ans, extrêmes 5-17 ans) atteints de gliomes diffus intrinsèques non disséminés du tronc cérébral nouvellement diagnostiqués et de gliomes de haut grade. Dans la partie recherche de dose de l'essai, les patients ont reçu de la capécitabine avec une radiothérapie concomitante à des doses allant de 500 mg/m2 à 850 mg/m2 toutes les 12 heures pendant 9 semaines maximum. Après une pause de 2 semaines, les patients ont reçu 1250 mg/m2 de capécitabine toutes les 12 heures les jours 1 à 14 d'un cycle de 21 jours pendant un maximum de 3 cycles. La dose maximale tolérée (DMT) de capécitabine administrée en concomitance avec la radiothérapie était de 650 mg/m2 toutes les 12 heures. Les principales toxicités limitant la dose étaient l'érythrodysesthésie palmo-plantaire et l'élévation de l'alanine aminotransférase (ALT).
Le deuxième essai a été mené chez 34 patients pédiatriques supplémentaires atteints de gliomes diffus intrinsèques non disséminés du tronc cérébral nouvellement diagnostiqués (âge médian 7 ans, intervalle 3-16 ans) et 10 patients pédiatriques qui ont reçu la DMT de capécitabine dans l'essai de recherche de dose et ont rencontré les critères d'éligibilité pour cet essai. Tous les patients ont reçu 650 mg/m2 de capécitabine toutes les 12 heures avec une radiothérapie concomitante pendant jusqu'à 9 semaines. Après une pause de 2 semaines, les patients ont reçu 1250 mg/m2 de capécitabine toutes les 12 heures les jours 1 à 14 d'un cycle de 21 jours pendant un maximum de 3 cycles.
Il n'y a eu aucune amélioration du taux de survie sans progression à un an et du taux de survie globale à un an chez les patients pédiatriques atteints de gliomes intrinsèques du tronc cérébral nouvellement diagnostiqués qui ont reçu de la capécitabine par rapport à une population similaire de patients pédiatriques qui ont participé à d'autres essais cliniques.
Le profil des effets indésirables de la capécitabine était cohérent avec le profil des effets indésirables connus chez les adultes, à l'exception des anomalies de laboratoire qui se sont produites plus fréquemment chez les patients pédiatriques. Les anomalies biologiques les plus fréquemment signalées (incidence par patient ≥ 40 %) étaient une augmentation des ALAT (75 %), une lymphocytopénie (73 %), une leucopénie (73 %), une hypokaliémie (68 %), une thrombocytopénie (57 %), une hypoalbuminémie (55 %). %), neutropénie (50 %), hématocrite bas (50 %), hypocalcémie (48 %), hypophosphatémie (45 %) et hyponatrémie (45 %).
Utilisation gériatrique
Les médecins doivent être particulièrement attentifs à la surveillance des effets indésirables de XELODA 500 mg chez la personne âgée [voir AVERTISSEMENTS ET PRECAUTIONS ].
Insuffisance hépatique
Soyez prudent lorsque des patients présentant une dysfonction hépatique légère à modérée due à des métastases hépatiques sont traités par XELODA. L'effet d'un dysfonctionnement hépatique sévère sur XELODA n'est pas connu [voir AVERTISSEMENTS ET PRECAUTIONS et PHARMACOLOGIE CLINIQUE ].
Insuffisance rénale
Les patients présentant une insuffisance rénale modérée (clairance de la créatinine = 30 à 50 ml/min) et sévère (clairance de la créatinine CONTRE-INDICATIONS , AVERTISSEMENTS ET PRECAUTIONS , DOSAGE ET ADMINISTRATION , et PHARMACOLOGIE CLINIQUE ].
SURDOSAGE
Les manifestations d'un surdosage aigu comprendraient des nausées, des vomissements, de la diarrhée, une irritation et des saignements gastro-intestinaux et une dépression de la moelle osseuse. La prise en charge médicale du surdosage doit inclure les interventions médicales de soutien habituelles visant à corriger les manifestations cliniques présentées. Bien qu'aucune expérience clinique utilisant la dialyse comme traitement d'un surdosage de XELODA 500 mg n'ait été rapportée, la dialyse peut être bénéfique pour réduire les concentrations circulantes de 5'-DFUR, un métabolite de faible poids moléculaire du composé parent.
Des doses uniques de XELODA 500 mg n'ont pas été mortelles pour les souris, les rats et les singes à des doses allant jusqu'à 2 000 mg/kg (2,4, 4,8 et 9,6 fois la dose quotidienne recommandée chez l'homme sur une base en mg/m2).
CONTRE-INDICATIONS
Insuffisance rénale sévère
XELODA est contre-indiqué chez les patients présentant une insuffisance rénale sévère (clairance de la créatinine inférieure à 30 mL/min [Cockroft et Gault]) [voir Utilisation dans des populations spécifiques et PHARMACOLOGIE CLINIQUE ].
Hypersensibilité
XELODA 500 mg est contre-indiqué chez les patients présentant une hypersensibilité connue à la capécitabine ou à l'un de ses composants. XELODA est contre-indiqué chez les patients présentant une hypersensibilité connue au 5fluorouracile.
PHARMACOLOGIE CLINIQUE
Mécanisme d'action
Les enzymes convertissent la capécitabine en 5-fluorouracile (5-FU) in vivo. Les cellules normales et tumorales métabolisent le 5-FU en 5-fluoro-2'-désoxyuridine monophosphate (FdUMP) et 5-fluorouridine triphosphate (FUTP). Ces métabolites provoquent des lésions cellulaires par deux mécanismes différents. Premièrement, le FdUMP et le cofacteur folate, le N5-10-méthylènetétrahydrofolate, se lient à la thymidylate synthase (TS) pour former un complexe ternaire lié par covalence. Cette liaison inhibe la formation de thymidylate à partir du 2'-désoxyuridylate. Le thymidylate est le précurseur nécessaire du triphosphate de thymidine, essentiel à la synthèse de l'ADN, de sorte qu'une carence en ce composé peut inhiber la division cellulaire. Deuxièmement, les enzymes transcriptionnelles nucléaires peuvent incorporer par erreur FUTP à la place de l'uridine triphosphate (UTP) lors de la synthèse de l'ARN. Cette erreur métabolique peut interférer avec le traitement de l'ARN et la synthèse des protéines.
Pharmacocinétique
Absorption
Après administration orale de 1255 mg/m2 BID à des patients cancéreux, la capécitabine a atteint des concentrations sanguines maximales en environ 1,5 heure (Tmax) avec des concentrations maximales de 5-FU survenant un peu plus tard, à 2 heures. La nourriture a réduit à la fois le taux et l'étendue de l'absorption de la capécitabine avec une Cmax et une ASC0-∞ moyennes diminuées de 60 % et 35 %, respectivement. La Cmax et l'ASC0-∞ du 5-FU ont également été réduites par la nourriture de 43 % et 21 %, respectivement. La nourriture a retardé le Tmax du parent et du 5-FU de 1,5 heure [voir AVERTISSEMENTS ET PRECAUTIONS , DOSAGE ET ADMINISTRATION , et Interaction médicament-aliment ].
La pharmacocinétique de XELODA 500 mg et de ses métabolites a été évaluée chez environ 200 patients cancéreux sur une plage posologique de 500 à 3500 mg/m2/jour. Dans cette plage, la pharmacocinétique de XELODA et de son métabolite, le 5'-DFCR, était proportionnelle à la dose et n'a pas changé avec le temps. Cependant, les augmentations des ASC du 5'-DFUR et du 5-FU étaient plus que proportionnelles à l'augmentation de la dose et l'ASC du 5-FU était supérieure de 34 % au jour 14 par rapport au jour 1. La variabilité interindividuelle des La Cmax et l'ASC du 5-FU étaient supérieures à 85 %.
Distribution
La liaison aux protéines plasmatiques de la capécitabine et de ses métabolites est inférieure à 60 % et n'est pas dépendante de la concentration. La capécitabine était principalement liée à l'albumine humaine (environ 35 %). XELODA 500 mg présente un faible potentiel d'interactions pharmacocinétiques liées à la liaison aux protéines plasmatiques.
Bioactivation et métabolisme
La capécitabine est largement métabolisée par voie enzymatique en 5-FU. Dans le foie, une carboxylestérase de 60 kDa hydrolyse une grande partie du composé en 5'-désoxy-5-fluorocytidine (5'-DFCR). La cytidine désaminase, une enzyme présente dans la plupart des tissus, y compris les tumeurs, convertit ensuite le 5'-DFCR en 5'-DFUR. L'enzyme, la thymidine phosphorylase (dThdPase), hydrolyse ensuite le 5'DFUR en le médicament actif 5-FU. De nombreux tissus du corps expriment la thymidine phosphorylase. Certains carcinomes humains expriment cette enzyme à des concentrations plus élevées que les tissus normaux environnants. Après administration orale de XELODA 7 jours avant l'intervention chirurgicale chez des patients atteints d'un cancer colorectal, le rapport médian de la concentration de 5-FU dans les tumeurs colorectales aux tissus adjacents était de 2,9 (intervalle de 0,9 à 8,0). Ces ratios n'ont pas été évalués chez des patientes atteintes d'un cancer du sein ni comparés à une perfusion de 5-FU.
Voie métabolique de la capécitabine en 5-FU
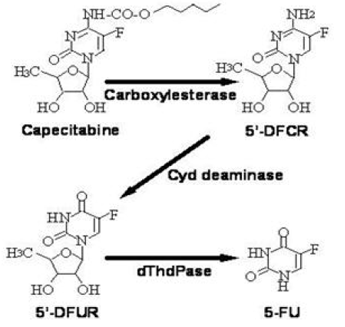
L'enzyme dihydropyrimidine déshydrogénase hydrogène le 5-FU, produit du métabolisme de la capécitabine, en 5-fluoro-5,6-dihydro-fluorouracile (FUH2), beaucoup moins toxique. La dihydropyrimidinase clive le cycle pyrimidine pour donner l'acide 5-fluoro-uréido-propionique (FUPA). Enfin, la β-uréido-propionase clive la FUPA en α-fluoro-β-alanine (FBAL) qui est éliminée dans l'urine.
Des études enzymatiques in vitro sur des microsomes hépatiques humains ont indiqué que la capécitabine et ses métabolites (5'-DFUR, 5'-DFCR, 5-FU et FBAL) n'inhibaient pas le métabolisme des substrats d'essai par les isoenzymes 1A2, 2A6, 3A4 du cytochrome P450, 2C19, 2D6 et 2E1.
Excrétion
La capécitabine et ses métabolites sont principalement excrétés dans l'urine ; 95,5 % de la dose de capécitabine administrée est récupérée dans les urines. L'excrétion fécale est minime (2,6 %). Le principal métabolite excrété dans les urines est le FBAL qui représente 57 % de la dose administrée. Environ 3 % de la dose administrée est excrétée dans l'urine sous forme inchangée. La demi-vie d'élimination de la capécitabine mère et du 5-FU était d'environ 0,75 heure.
Effet de l'âge, du sexe et de la race sur la pharmacocinétique de la capécitabine
Une analyse de population des données regroupées des deux grandes études contrôlées chez des patients atteints d'un cancer colorectal métastatique (n = 505) qui ont reçu XELODA 500 mg à 1250 mg/m2 deux fois par jour a indiqué que le sexe (202 femmes et 303 hommes) et la race (455 patients blancs/caucasiens, 22 patients noirs et 28 patients d'une autre race) n'ont aucune influence sur la pharmacocinétique du 5'DFUR, du 5-FU et du FBAL. L'âge n'a pas d'influence significative sur la pharmacocinétique du 5'-DFUR et du 5-FU entre 27 et 86 ans. Une augmentation de 20 % de l'âge entraîne une augmentation de 15 % de l'ASC de la FBAL [voir AVERTISSEMENTS ET PRECAUTIONS et DOSAGE ET ADMINISTRATION ].
Après administration orale de 825 mg/m2 de capécitabine deux fois par jour pendant 14 jours, les patients japonais (n = 18) avaient une Cmax d'environ 36 % et une ASC de la capécitabine inférieures de 24 % à celles des patients caucasiens (n = 22). Les patients japonais avaient également une Cmax inférieure d'environ 25 % et une ASC 34 % inférieure pour le FBAL par rapport aux patients caucasiens. La signification clinique de ces différences est inconnue. Aucune différence significative n'a été observée dans l'exposition aux autres métabolites (5'-DFCR, 5'-DFUR et 5FU).
Effet de l'insuffisance hépatique
XELODA a été évalué chez 13 patients présentant une insuffisance hépatique légère à modérée due à des métastases hépatiques définies par un score composite comprenant la bilirubine, l'AST/ALT et la phosphatase alcaline après une dose unique de 1255 mg/m2 de XELODA. L'ASC0-∞ et la Cmax de la capécitabine ont toutes deux augmenté de 60 % chez les patients présentant un dysfonctionnement hépatique par rapport aux patients présentant une fonction hépatique normale (n = 14). L'ASC0-∞ et la Cmax du 5-FU n'ont pas été affectées. Chez les patients présentant une insuffisance hépatique légère à modérée due à des métastases hépatiques, la prudence s'impose lors de l'administration de XELODA 500 mg. L'effet d'un dysfonctionnement hépatique sévère sur XELODA 500 mg n'est pas connu [voir AVERTISSEMENTS ET PRECAUTIONS et Utilisation dans des populations particulières ].
Effet de l'insuffisance rénale
Après administration orale de 1250 mg/m2 de capécitabine deux fois par jour à des patients cancéreux présentant divers degrés d'insuffisance rénale, les patients présentant une insuffisance rénale modérée (clairance de la créatinine = 30 à 50 ml/min) et sévère (clairance de la créatinine 80 mL/min). L'exposition systémique au 5'-DFUR était respectivement de 42 % et 71 % plus élevée chez les insuffisants rénaux modérés et sévères que chez les patients normaux. L'exposition systémique à la capécitabine était supérieure d'environ 25 % chez les patients atteints d'insuffisance rénale modérée et sévère [voir DOSAGE ET ADMINISTRATION , CONTRE-INDICATIONS , AVERTISSEMENTS ET PRECAUTIONS , et Utilisation dans des populations particulières ].
Effet de la capécitabine sur la pharmacocinétique de la warfarine
Chez quatre patients atteints de cancer, l'administration chronique de capécitabine (1 250 mg/m2 bid) avec une dose unique de 20 mg de warfarine a augmenté l'ASC moyenne de la S-warfarine de 57 % et a diminué sa clairance de 37 %. L'ASC corrigée à l'inclusion de l'INR chez ces 4 patients a été multipliée par 2,8 et la valeur moyenne maximale de l'INR observée a augmenté de 91 % [voir AVERTISSEMENT DE BOÎTE et INTERACTIONS MÉDICAMENTEUSES ].
Effet des antiacides sur la pharmacocinétique de la capécitabine
Lorsque Maalox® (20 mL), un antiacide contenant de l'hydroxyde d'aluminium et de l'hydroxyde de magnésium, a été administré immédiatement après XELODA (1250 mg/m2, n = 12 patients atteints de cancer), l'ASC et la Cmax ont augmenté de 16 % et 35 %, respectivement, pour la capécitabine et de 18 % et 22 %, respectivement, pour le 5'-DFCR. Aucun effet n'a été observé sur les trois autres métabolites majeurs (5'-DFUR, 5-FU, FBAL) de XELODA.
Effet de l'allopurinol sur la capécitabine
La littérature publiée a rapporté que l'utilisation concomitante avec l'allopurinol peut diminuer la conversion de la capécitabine en métabolites actifs, FdUMP et FUTP ; cependant, la signification clinique n'a pas été entièrement caractérisée.
Effet de la capécitabine sur la pharmacocinétique du docétaxel et vice versa
Une étude de phase 1 a évalué l'effet de XELODA sur la pharmacocinétique du docétaxel (Taxotere®) et l'effet du docétaxel sur la pharmacocinétique de XELODA a été menée chez 26 patients atteints de tumeurs solides. XELODA s'est avéré n'avoir aucun effet sur la pharmacocinétique du docétaxel (Cmax et ASC) et le docétaxel n'a aucun effet sur la pharmacocinétique de la capécitabine et du précurseur du 5-FU, le 5'-DFUR.
Etudes cliniques
Cancer du côlon adjuvant
Un essai clinique multicentrique randomisé et contrôlé de phase 3 chez des patients atteints d'un cancer du côlon Dukes' C (X-ACT) a fourni des données concernant l'utilisation de XELODA pour le traitement adjuvant des patients atteints d'un cancer du côlon. L'objectif principal de l'étude était de comparer la survie sans maladie (DFS) chez les patients recevant XELODA à ceux recevant IV 5-FU/LV seul. Dans cet essai, 1987 patients ont été randomisés soit pour recevoir XELODA 1250 mg/m2 par voie orale deux fois par jour pendant 2 semaines suivi d'une période de repos d'une semaine, administrés sous forme de cycles de 3 semaines pour un total de 8 cycles (24 semaines) ou IV bolus 5-FU 425 mg/m2 et 20 mg/m2 IV de leucovorine les jours 1 à 5, administrés en cycles de 4 semaines pour un total de 6 cycles (24 semaines). Les patients de l'étude devaient être âgés de 18 à 75 ans avec un cancer du côlon de stade C de Dukes confirmé histologiquement avec au moins un ganglion lymphatique positif et avoir subi (dans les 8 semaines précédant la randomisation) une résection complète de la tumeur primaire sans preuve macroscopique ou microscopique de tumeur restante. Les patients devaient également n'avoir subi aucune chimiothérapie ou immunothérapie cytotoxique antérieure (à l'exception des stéroïdes) et avoir un indice de performance ECOG de 0 ou 1 (KPS ≥ 70 %), ANC ≥ 1,5 x 109/L, plaquettes ≥ 100 x 109/L, créatinine sérique ≤ 1,5 LSN, bilirubine totale ≤ 1,5 LSN, AST/ALT ≤ 2,5 LSN et ACE dans les limites normales au moment de la randomisation.
Les données démographiques initiales pour les patients XELODA et 5-FU/LV sont présentées dans le tableau 10. Les caractéristiques initiales étaient bien équilibrées entre les bras.
Tous les patients ayant une fonction rénale normale ou une insuffisance rénale légère ont commencé le traitement à la dose initiale complète de 1250 mg/m2 par voie orale deux fois par jour. La dose initiale a été réduite chez les patients présentant une insuffisance rénale modérée (clairance de la créatinine calculée de 30 à 50 ml/min) au départ [voir DOSAGE ET ADMINISTRATION ]. Par la suite, pour tous les patients, les doses ont été ajustées au besoin en fonction de la toxicité. La gestion de la dose de XELODA 500 mg comprenait des réductions de dose, des retards de cycle et des interruptions de traitement (voir tableau 11).
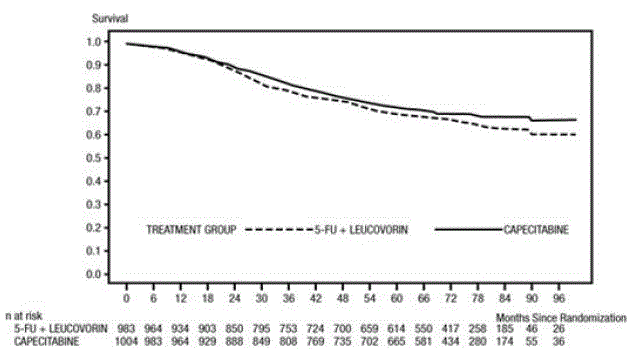
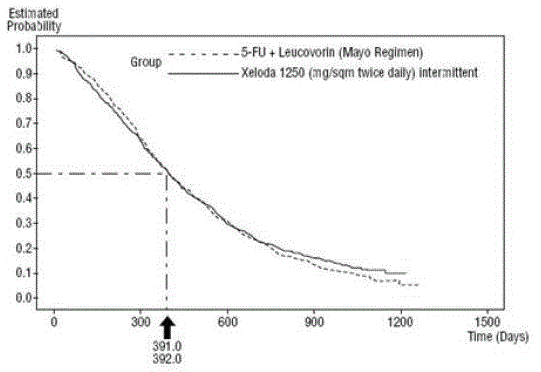
Le suivi médian au moment de l'analyse était de 83 mois (6,9 ans). Le risque relatif de SSM pour XELODA 500 mg par rapport au 5-FU/LV était de 0,88 (IC à 95 % 0,77 – 1,01) (voir et Figure 1 ). Étant donné que la limite de confiance bilatérale supérieure à 95 % du rapport de risque était inférieure à 1,20, XELODA était non inférieur au 5-FU/LV. Le choix de la marge de non-infériorité de 1,20 correspond au maintien d'environ 75% de l'effet 5-FU/LV sur la SSM. Le risque relatif de XELODA par rapport au 5-FU/LV en ce qui concerne la survie globale était de 0,86 (IC à 95 % 0,74 – 1,01). Les taux de survie globale à 5 ans étaient de 71,4 % pour XELODA et de 68,4 % pour le 5-FU/LV (voir Figure 2).
Figure 1 Estimations de Kaplan-Meier de la survie sans maladie (toute population randomisée)a
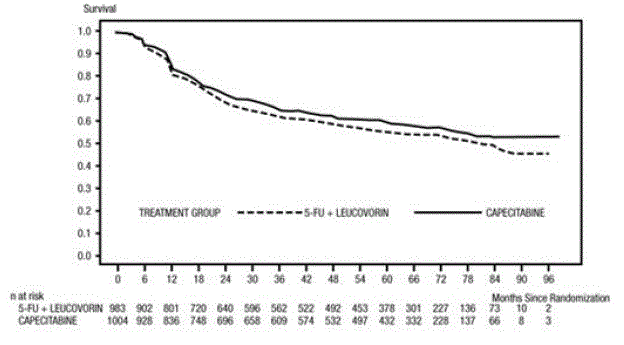
Figure 2 Estimations de Kaplan-Meier de la survie globale (toute population randomisée)
Cancer colorectal métastatique
Général
La dose recommandée de XELODA 500 mg a été déterminée dans une étude clinique randomisée, en ouvert, explorant l'efficacité et l'innocuité d'un traitement continu par la capécitabine (1331 mg/m2/jour en deux doses fractionnées, n = 39), d'un traitement intermittent par la capécitabine ( 2 510 mg/m2/jour en deux prises, n=34), et un traitement intermittent par la capécitabine en association avec la leucovorine (LV) orale (capécitabine 1 657 mg/m2/jour en deux prises, n=35 ; leucovorine 60 mg/ jour) chez les patients atteints d'un carcinome colorectal avancé et/ou métastatique dans le cadre métastatique de première ligne. Il n'y avait aucun avantage apparent dans le taux de réponse à l'ajout de leucovorine à XELODA ; cependant, la toxicité a été augmentée. XELODA, 1250 mg/m2 deux fois par jour pendant 14 jours suivis d'une semaine de repos, a été sélectionné pour un développement clinique plus poussé sur la base du profil global d'innocuité et d'efficacité des trois schémas étudiés.
Monothérapie
Les données de deux essais cliniques ouverts, multicentriques, randomisés et contrôlés portant sur 1207 patients appuient l'utilisation de XELODA dans le traitement de première intention des patients atteints d'un carcinome colorectal métastatique. Les deux études cliniques étaient de conception identique et ont été menées dans 120 centres de différents pays. L'étude 1 a été menée aux États-Unis, au Canada, au Mexique et au Brésil ; L'étude 2 a été menée en Europe, en Israël, en Australie, en Nouvelle-Zélande et à Taïwan. Au total, dans les deux essais, 603 patients ont été randomisés pour recevoir un traitement par XELODA 500 mg à une dose de 1250 mg/m2 deux fois par jour pendant 2 semaines suivi d'une période de repos d'une semaine et administré sous forme de cycles de 3 semaines ; 604 patients ont été randomisés pour recevoir un traitement par 5-FU et leucovorine (20 mg/m2 de leucovorine IV suivi de 425 mg/m2 bolus IV de 5-FU, les jours 1 à 5, tous les 28 jours).
Dans les deux essais, la survie globale, le temps jusqu'à progression et le taux de réponse (réponses complètes et partielles) ont été évalués. Les réponses ont été définies par les critères de l'Organisation mondiale de la santé et soumises à un comité d'examen indépendant (IRC) en aveugle. Les différences d'évaluation entre l'investigateur et l'IRC ont été réconciliées par le promoteur, en aveugle au bras de traitement, selon un algorithme spécifié. La survie a été évaluée sur la base d'une analyse de non-infériorité.
Les données démographiques de base pour les patients XELODA et 5-FU/LV sont présentées dans le tableau 13.
Les paramètres d'efficacité pour les deux essais de phase 3 sont présentés dans et
Figure 3 Courbe de Kaplan-Meier pour la survie globale des données regroupées (études 1 et 2)
XELODA a été supérieur au 5-FU/LV pour le taux de réponse objective dans les études 1 et 2. La similarité de XELODA 500 mg et 5-FU/LV dans ces études a été évaluée en examinant la différence potentielle entre les deux traitements. Afin de s'assurer que XELODA 500 mg a un effet de survie cliniquement significatif, des analyses statistiques ont été effectuées pour déterminer le pourcentage de l'effet de survie du 5-FU/LV qui a été retenu par XELODA. L'estimation de l'effet de survie du 5-FU/LV a été dérivée d'une méta-analyse de dix études randomisées de la littérature publiée comparant le 5-FU à des régimes de 5-FU/LV qui étaient similaires aux bras témoins utilisés dans ces études. 1 et 2. La méthode de comparaison des traitements a consisté à examiner le pire cas (limite supérieure de confiance à 95 %) pour la différence entre le 5-FU/LV et XELODA 500 mg, et à montrer que la perte de plus de 50 % du 5- L'effet de survie FU/LV a été exclu. Il a été démontré que le pourcentage de l'effet de survie du 5-FU/LV maintenu était d'au moins 61 % pour l'étude 2 et de 10 % pour l'étude 1. Le résultat combiné est cohérent avec une rétention d'au moins 50 % de l'effet de 5 -FU/VG. Il est à noter que ces valeurs d'effet préservé sont basées sur la borne supérieure de la différence 5-FU/LV vs XELODA 500mg. Ces résultats n'excluent pas la possibilité d'une véritable équivalence de XELODA 500mg au 5-FU/LV (voir et figure 3 ).
Cancer du sein
XELODA a été évalué dans des essais cliniques en association avec le docétaxel (Taxotere®) et en monothérapie.
En association avec le docétaxel
La dose de XELODA utilisée dans l'essai clinique de phase 3 en association avec le docétaxel était basée sur les résultats d'une étude de phase 1, où une gamme de doses de docétaxel administrées en cycles de 3 semaines en association avec un régime intermittent de XELODA (14 jours de traitement, suivi d'une période de repos de 7 jours) ont été évalués. Le schéma posologique combiné a été sélectionné en fonction du profil de tolérance des 75 mg/m2 administrés en cycles de 3 semaines de docétaxel en association avec 1250 mg/m2 deux fois par jour pendant 14 jours de XELODA 500 mg administrés en cycles de 3 semaines. La dose approuvée de 100 mg/m2 de docétaxel administrée en cycles de 3 semaines était le groupe témoin de l'étude de phase 3.
XELODA en association avec le docétaxel a été évalué dans le cadre d'un essai ouvert, multicentrique et randomisé dans 75 centres en Europe, en Amérique du Nord, en Amérique du Sud, en Asie et en Australie. Un total de 511 patientes atteintes d'un cancer du sein métastatique résistant à, ou récidivant pendant ou après un traitement contenant des anthracyclines, ou rechutant pendant ou récidivant dans les 2 ans suivant la fin d'un traitement adjuvant contenant des anthracyclines ont été recrutées. Deux cent cinquante-cinq (255) patients ont été randomisés pour recevoir XELODA 1250 mg/m2 deux fois par jour pendant 14 jours suivis d'une semaine sans traitement et du docétaxel 75 mg/m2 en perfusion intraveineuse d'une heure administré en cycles de 3 semaines. Dans le bras monothérapie, 256 patients ont reçu du docétaxel 100 mg/m2 en perfusion intraveineuse d'une heure administrée en cycles de 3 semaines. Les caractéristiques démographiques des patients sont fournies dans le tableau 16.
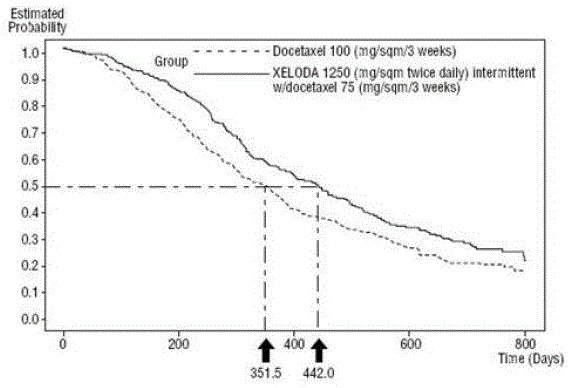
XELODA en association avec le docétaxel a entraîné une amélioration statistiquement significative du temps jusqu'à la progression de la maladie, de la survie globale et du taux de réponse objective par rapport à la monothérapie avec le docétaxel, comme indiqué dans et Figure 5 .
Figure 4 Estimations de Kaplan-Meier du délai avant progression de la maladie XELODA et docétaxel par rapport au docétaxel
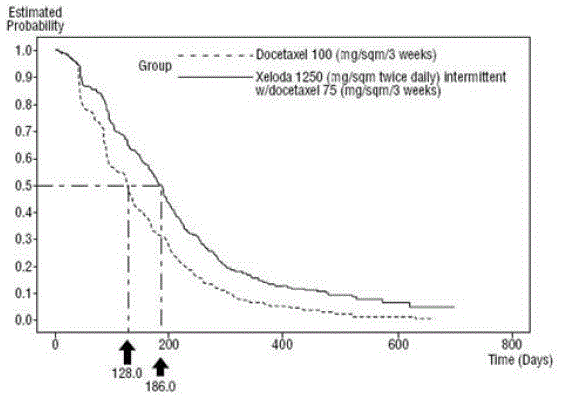
Figure 5 Estimations de Kaplan-Meier de la survie de XELODA et du docétaxel par rapport au docétaxel
Monothérapie
L'activité antitumorale de XELODA en monothérapie a été évaluée dans un essai ouvert à un seul bras mené dans 24 centres aux États-Unis et au Canada. Au total, 162 patientes atteintes d'un cancer du sein de stade IV ont été recrutées. Le critère d'évaluation principal était le taux de réponse tumorale chez les patients atteints d'une maladie mesurable, la réponse étant définie comme une diminution ≥ 50 % de la somme des produits des diamètres perpendiculaires de la maladie mesurable de manière bidimensionnelle pendant au moins 1 mois. XELODA a été administré à une dose de 1 255 mg/m2 deux fois par jour pendant 2 semaines, suivie d'une période de repos d'une semaine et administré en cycles de 3 semaines. Les caractéristiques démographiques et cliniques de base pour tous les patients (n = 162) et ceux atteints d'une maladie mesurable (n = 135) sont présentées dans . La résistance a été définie comme une maladie évolutive pendant le traitement, avec ou sans réponse initiale, ou une rechute dans les 6 mois suivant la fin du traitement avec une chimiothérapie adjuvante contenant de l'anthracycline.
Les réponses antitumorales pour les patients atteints d'une maladie résistante à la fois au paclitaxel et à une anthracycline sont présentées dans
Pour le sous-groupe de 43 patients doublement résistants, le délai médian jusqu'à progression était de 102 jours et la survie médiane était de 255 jours. Le taux de réponse objective dans cette population a été étayé par un taux de réponse de 18,5 % (1 RC, 24 RP) dans la population globale de 135 patients atteints d'une maladie mesurable, qui étaient moins résistants à la chimiothérapie (voir ). Le délai médian jusqu'à progression était de 90 jours et la survie médiane était de 306 jours.
INFORMATIONS PATIENTS
XELODA® (zeh-LOE-duh) (capécitabine) comprimés
Quelle est l'information la plus importante que je devrais connaître sur XELODA 500 mg ?
XELODA 500 mg peut provoquer des effets indésirables graves, notamment :
- XELODA peut interagir avec des anticoagulants, comme la warfarine (COUMADIN®). La prise de XELODA 500 mg avec ces médicaments peut modifier la vitesse de coagulation de votre sang et provoquer des saignements pouvant entraîner la mort. Cela peut se produire quelques jours seulement après le début de la prise de XELODA 500 mg, ou plus tard pendant le traitement, et éventuellement même dans le mois qui suit l'arrêt de la prise de XELODA. Votre risque peut être plus élevé parce que vous avez un cancer et si vous avez plus de 60 ans.
- Avant de prendre XELODA, informez votre fournisseur de soins de santé si vous prenez de la warfarine (COUMADIN) ou un autre anticoagulant.
- Si vous prenez de la warfarine (COUMADIN) ou un autre anticoagulant semblable à la warfarine (COUMADIN) pendant le traitement par XELODA, votre fournisseur de soins de santé doit effectuer régulièrement des analyses de sang pour vérifier la vitesse à laquelle votre sang coagule pendant et après l'arrêt du traitement par XELODA. Votre fournisseur de soins de santé peut modifier votre dose de médicament anticoagulant si nécessaire.
Voir "Quels sont les effets secondaires possibles de XELODA 500mg ?" pour plus d'informations sur les effets secondaires.
Qu'est-ce que XELODA ?
XELODA est un médicament délivré sur ordonnance utilisé pour traiter les personnes atteintes de :
- cancer du côlon qui s'est propagé aux ganglions lymphatiques de la région proche du côlon (stade C de Dukes), après une intervention chirurgicale.
- cancer du côlon ou du rectum (colorectal) qui s'est propagé à d'autres parties du corps (métastatique), comme premier traitement de votre cancer à ce stade.
- cancer du sein qui s'est propagé à d'autres parties du corps (métastatique) avec un autre médicament appelé docétaxel après un traitement avec certains autres médicaments anticancéreux n'a pas fonctionné.
- cancer du sein qui s'est propagé à d'autres parties du corps et qui ne s'est pas amélioré après un traitement par le paclitaxel et certains autres médicaments anticancéreux, ou qui ne peut plus recevoir de traitement avec certains médicaments anticancéreux.
On ne sait pas si XELODA 500 mg est sûr et efficace chez les enfants.
Ne prenez jamais XELODA si vous :
- avoir de graves problèmes rénaux
- êtes allergique à la capécitabine, au 5-fluorouracile ou à l'un des ingrédients contenus dans XELODA. Voir la fin de cette notice pour une liste complète des ingrédients de XELODA.
Adressez-vous à votre fournisseur de soins de santé avant de prendre XELODA si vous n'êtes pas sûr d'avoir l'une des conditions énumérées ci-dessus.
Avant de prendre XELODA, informez votre fournisseur de soins de santé de toutes vos conditions médicales, y compris si vous :
Voir « Quelle est l'information la plus importante que je devrais connaître sur XELODA ? »
- ont eu des problèmes cardiaques
- avez des problèmes rénaux ou hépatiques
- on vous a dit que vous manquiez de l'enzyme DPD (dihydropyrimidine déshydrogénase).
- êtes enceinte ou envisagez de devenir enceinte. XELODA 500 mg peut nuire à votre bébé à naître. Votre fournisseur de soins de santé devrait faire un test de grossesse avant de commencer le traitement par XELODA. Informez immédiatement votre fournisseur de soins de santé si vous tombez enceinte ou si vous pensez être enceinte pendant le traitement par XELODA.
- Femelles qui sont en mesure de devenir enceintes doivent utiliser une contraception efficace pendant le traitement et pendant 6 mois après la dernière dose. Parlez à votre fournisseur de soins de santé des choix de contraception qui pourraient vous convenir pendant le traitement par XELODA.
- Mâles qui ont des partenaires féminines capables de devenir enceintes doivent utiliser une contraception efficace pendant le traitement et pendant 3 mois après la dernière dose.
- allaitez ou envisagez d'allaiter. On ne sait pas si XELODA passe dans votre lait maternel. Ne pas allaiter pendant le traitement par XELODA 500 mg et pendant 2 semaines après la dernière dose.
Informez votre fournisseur de soins de santé de tous les médicaments que vous prenez, y compris les médicaments sur ordonnance et en vente libre, les vitamines et les suppléments à base de plantes. XELODA peut modifier le mode d'action d'autres médicaments, et d'autres médicaments peuvent modifier le mode d'action de XELODA.
Connaissez les médicaments que vous prenez. Conservez-en une liste pour la montrer à votre fournisseur de soins de santé et à votre pharmacien lorsque vous recevez un nouveau médicament.
Comment dois-je prendre XELODA 500 mg ?
- Prenez XELODA exactement comme votre professionnel de la santé vous a dit de le prendre.
- Votre fournisseur de soins de santé vous dira combien de XELODA prendre et quand le prendre.
- Prenez XELODA 2 fois par jour, 1 fois le matin et 1 fois le soir.
- Prenez XELODA 500 mg dans les 30 minutes suivant la fin d'un repas.
- Avalez les comprimés de XELODA 500mg entiers avec de l'eau. Ne pas écraser ou couper les comprimés de XELODA 500mg. Si vous ne pouvez pas avaler les comprimés de XELODA entiers, informez-en votre médecin.
- Votre fournisseur de soins de santé peut modifier votre dose, arrêter temporairement ou arrêter définitivement le traitement par XELODA 500 mg si vous développez des effets indésirables.
- Si vous avez pris trop de XELODA, appelez votre fournisseur de soins de santé ou rendez-vous immédiatement aux urgences de l'hôpital le plus proche.
Quels sont les effets secondaires possibles de XELODA ?
XELODA peut provoquer des effets indésirables graves, notamment :
Les nausées et les vomissements sont fréquents avec XELODA. Si vous perdez l'appétit, si vous vous sentez faible et si vous avez des nausées, des vomissements ou de la diarrhée, vous pouvez rapidement vous déshydrater.
Arrêtez de prendre XELODA et appelez immédiatement votre fournisseur de soins de santé si vous :
Si votre nombre de globules blancs est très bas, vous courez un risque accru d'infection. Appelez immédiatement votre fournisseur de soins de santé si vous développez une fièvre de 100,5 oF ou plus ou si vous présentez d'autres signes et symptômes d'infection.
- Voir « Quelle est l'information la plus importante que je devrais connaître sur XELODA ? »
- Diarrhée. La diarrhée est fréquente avec XELODA et peut parfois être sévère. Arrêtez de prendre XELODA et appelez immédiatement votre fournisseur de soins de santé si le nombre de selles que vous avez au cours d'une journée augmente de 4 selles ou plus que d'habitude pour vous ou des selles nocturnes. Demandez à votre fournisseur de soins de santé quels médicaments vous pouvez prendre pour traiter votre diarrhée. Si vous avez une diarrhée sanglante sévère avec des douleurs abdominales sévères et de la fièvre, arrêtez de prendre Xeloda 500 mg et appelez votre médecin ou rendez-vous immédiatement aux urgences de l'hôpital le plus proche.
- Problèmes cardiaques. XELODA peut provoquer des problèmes cardiaques, notamment : crise cardiaque et diminution du flux sanguin vers le cœur, douleurs thoraciques, battements cardiaques irréguliers, modifications de l'activité électrique de votre cœur observées sur un électrocardiogramme (ECG), problèmes de muscle cardiaque, insuffisance cardiaque et décès. Arrêtez de prendre XELODA 500 mg et appelez votre professionnel de la santé ou rendez-vous immédiatement aux urgences de l'hôpital le plus proche si vous présentez de nouveaux symptômes d'un problème cardiaque, notamment :
- douleur thoracique
- vertiges
- essoufflement
- étourdissement
- Perte excessive de liquide corporel (déshydratation) et insuffisance rénale. Une déshydratation peut survenir avec XELODA 500 mg et peut provoquer une insuffisance rénale soudaine pouvant entraîner la mort. Vous êtes plus à risque si vous avez des problèmes rénaux avant de prendre XELODA 500 mg et si vous prenez également d'autres médicaments pouvant causer des problèmes rénaux.
- vomir 2 fois ou plus par jour.
- ne peuvent manger ou boire qu'un peu de temps en temps, ou pas du tout à cause des nausées.
- avoir la diarrhée. Voir « diarrhée » ci-dessus.
- Réactions sévères de la peau et de la bouche.
- XELODA 500 mg peut provoquer des réactions cutanées sévères pouvant entraîner la mort. Informez immédiatement votre fournisseur de soins de santé si vous développez une éruption cutanée, une cloque et une desquamation de votre peau. Votre fournisseur de soins de santé peut vous dire d'arrêter de prendre XELODA si vous présentez une réaction cutanée grave. Ne prenez plus XELODA 500 mg si cela se produit.
- XELODA 500 mg peut également provoquer le syndrome « mains et pieds ». Le syndrome des mains et des pieds est courant avec XELODA 500 mg et peut provoquer un engourdissement et des changements de sensation dans les mains et les pieds, ou provoquer des rougeurs, des douleurs, un gonflement des mains et des pieds. Arrêtez de prendre XELODA et appelez immédiatement votre fournisseur de soins de santé si vous présentez l'un de ces symptômes et que vous n'êtes pas en mesure de vaquer à vos activités habituelles.
- Le syndrome des mains et des pieds peut entraîner une perte d'empreintes digitales qui pourrait avoir un impact sur votre identification.
- vous pouvez avoir des plaies dans la bouche ou sur la langue lorsque vous prenez XELODA. Arrêtez de prendre XELODA 500 mg et appelez votre professionnel de la santé si vous présentez une rougeur douloureuse, un gonflement ou des ulcères dans la bouche ou la langue, ou si vous avez des difficultés à manger.
- Augmentation du niveau de bilirubine dans vos problèmes sanguins et hépatiques. Une augmentation de la bilirubine dans votre sang est courante avec XELODA 500 mg et peut aussi parfois être sévère. Votre fournisseur de soins de santé vérifiera si vous avez ces problèmes pendant le traitement par XELODA.
- Diminution du nombre de globules blancs, de plaquettes et de globules rouges. Votre fournisseur de soins de santé effectuera des tests sanguins pendant le traitement par XELODA pour vérifier votre nombre de cellules sanguines.
Les personnes de 80 ans ou plus peuvent être plus susceptibles de développer des effets secondaires graves ou graves avec XELODA.
Les effets indésirables les plus courants de XELODA 500 mg incluent :
- diarrhée
- douleur dans la région de l'estomac (abdominale)
- syndrome des mains et des pieds
- faiblesse et fatigue
- nausée
- vomi
- augmentation des quantités de produits de dégradation des globules rouges (bilirubine dans votre sang
Des réactions allergiques graves peuvent survenir avec XELODA. Informez votre fournisseur de soins de santé si vous avez déjà eu une réaction allergique à la capécitabine ou au 5-fluorouracile. Voir « Ne prenez pas XELODA si vous : ». Arrêtez de prendre XELODA 500 mg et appelez immédiatement votre professionnel de la santé ou rendez-vous aux urgences si vous présentez l'un des symptômes suivants d'une réaction allergique grave :
- démangeaisons rouges sur la peau (urticaire)
- rougeur de la peau
- gonflement du visage, des lèvres, de la langue ou de la gorge
- éruption
- démangeaison
- difficulté à avaler ou à respirer
XELODA 500 mg peut entraîner des problèmes de fertilité chez les femmes et les hommes. Cela peut affecter la capacité d'avoir un enfant. Parlez à votre fournisseur de soins de santé si vous avez des inquiétudes au sujet de la fertilité.
Ce ne sont pas tous les effets secondaires possibles de XELODA.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
Comment dois-je conserver XELODA ?
- Conservez XELODA 500 mg à température ambiante entre 68 °F et 77 °F (20 °C et 25 °C).
- Conservez XELODA 500 mg dans un récipient hermétiquement fermé.
- Demandez à votre fournisseur de soins de santé ou à votre pharmacien comment jeter en toute sécurité tout XELODA inutilisé.
Gardez XELODA et tous les médicaments hors de la portée des enfants.
Informations générales sur l'utilisation sûre et efficace de XELODA.
Les médicaments sont parfois prescrits à des fins autres que celles énumérées dans une notice d'information destinée aux patients. N'utilisez pas XELODA pour une affection pour laquelle il n'a pas été prescrit. Ne donnez pas XELODA 500 mg à d'autres personnes, même si elles présentent les mêmes symptômes que vous. Cela peut leur nuire. Vous pouvez demander à votre pharmacien ou à votre professionnel de la santé des informations sur XELODA 500 mg destinées aux professionnels de la santé.
Quels sont les ingrédients de XELODA 500 mg ?
Ingrédient actif: capécitabine
Ingrédients inactifs: lactose anhydre, croscarmellose sodique, hydroxypropylméthylcellulose, cellulose microcristalline, stéarate de magnésium et eau purifiée. Le pelliculage pêche ou pêche clair contient de l'hydroxypropylméthylcellulose, du talc, du dioxyde de titane et des oxydes de fer jaune et rouge synthétiques.
Pour plus d'informations, rendez-vous sur http://www.gene.com/patients/medicines/xeloda 500mg ou appelez le 1-877-436-3683.
Ces informations destinées aux patients ont été approuvées par la Food and Drug Administration des États-Unis.