Traitement pour les cheveux: Avodart 0.5mg Dutasteride Utilisations, effets secondaires et dosage. Prix en Pharmacie. Medicaments generiques sans ordonnance.
Qu'est-ce qu'Avodart 0,5 mg et comment est-il utilisé ?
Avodart 0,5 mg est un médicament délivré sur ordonnance utilisé pour traiter les symptômes d'une hypertrophie de la prostate (hyperplasie bénigne de la prostate). Avodart peut être utilisé seul ou avec d'autres médicaments.
Avodart 0,5 mg appartient à une classe de médicaments appelés inhibiteurs de la 5-alpha-réductase.
On ne sait pas si Avodart 0,5 mg est sûr et efficace chez les enfants.
Quels sont les effets secondaires d'Avodart ?
Les effets secondaires d'Avodart 0,5 mg incluent :
- urticaire,
- difficulté à respirer,
- gonflement du visage ou de la gorge,
- fièvre,
- mal de gorge,
- Yeux brûlants,
- douleurs cutanées et
- éruption cutanée rouge ou violette avec cloques et desquamation
Consultez immédiatement un médecin si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires les plus courants d'Avodart 0,5 mg incluent :
- diminution de la libido (libido),
- diminution de la quantité de sperme libérée pendant les rapports sexuels,
- impuissance (difficulté à obtenir ou à maintenir une érection), et
- sensibilité ou élargissement des seins
Dites au médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles d'Avodart. Pour plus d'informations, consultez votre médecin ou votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
LA DESCRIPTION
AVODART 0,5 mg est un composé 4-azastéroïde synthétique qui est un inhibiteur sélectif des isoformes de type 1 et de type 2 de la stéroïde 5 alpha-réductase, une enzyme intracellulaire qui convertit la testostérone en DHT.
Le dutastéride est chimiquement désigné par (5α,17β)-N-{2,5 bis(trifluorométhyl)phényl}-3-oxo-4-azaandrost-1-ène-17-carboxamide. La formule empirique du dutastéride est C27H30F6N2O2, représentant un poids moléculaire de 528,5 avec la formule structurelle suivante :
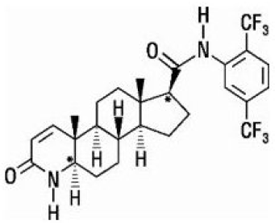
Le dutastéride est une poudre blanche à jaune pâle avec un point de fusion de 242° à 250°C. Il est soluble dans l'éthanol (44 mg/mL), le méthanol (64 mg/mL) et le polyéthylène glycol 400 (3 mg/mL), mais il est insoluble dans l'eau.
Chaque capsule de gélatine molle AVODART 0,5 mg, administrée par voie orale, contient 0,5 mg de dutastéride dissous dans un mélange de mono-di-glycérides d'acide caprylique/caprique et d'hydroxytoluène butylé. Les excipients inactifs contenus dans l'enveloppe de la gélule sont l'oxyde ferrique (jaune), la gélatine (provenant de sources bovines certifiées exemptes d'ESB), la glycérine et le dioxyde de titane. Les capsules de gélatine molle sont imprimées avec de l'encre rouge comestible.
LES INDICATIONS
Monothérapie
Les capsules de gélatine molle AVODART (dutastéride) sont indiquées pour le traitement de l'hyperplasie bénigne de la prostate (HBP) symptomatique chez les hommes présentant une hypertrophie de la prostate pour :
- améliorer les symptômes,
- réduire le risque de rétention urinaire aiguë (RAU) et
- réduire le risque de nécessiter une intervention chirurgicale liée à l'HBP.
Association avec un antagoniste alpha-adrénergique
AVODART en association avec l'antagoniste alpha-adrénergique, la tamsulosine, est indiqué pour le traitement de l'HBP symptomatique chez les hommes présentant une hypertrophie de la prostate.
Limites d'utilisation
AVODART 0,5 mg n'est pas approuvé pour la prévention du cancer de la prostate.
DOSAGE ET ADMINISTRATION
Les gélules doivent être avalées entières et non mâchées ni ouvertes, car le contact avec le contenu de la gélule peut entraîner une irritation de la muqueuse oropharyngée. AVODART peut être administré avec ou sans aliments.
Monothérapie
La dose recommandée d'AVODART est de 1 gélule (0,5 mg) une fois par jour.
Association avec un antagoniste alpha-adrénergique
La dose recommandée d'AVODART 0,5 mg est de 1 gélule (0,5 mg) une fois par jour et de 0,4 mg de tamsulosine une fois par jour.
COMMENT FOURNIE
Formes posologiques et points forts
Gélules de 0,5 mg, opaques, jaune terne, en gélatine, portant l'inscription « GX CE2 » à l'encre rouge sur une face.
Stockage et manutention
AVODART Les capsules de gélatine molle à 0,5 mg sont des capsules de gélatine oblongues, opaques, jaune terne, portant l'inscription « GX CE2 » avec de l'encre alimentaire rouge sur une face, conditionnées en flacons de 30 ( CDN 0173-0712-15) et 90 ( CDN 0173-0712-04) avec fermetures à l'épreuve des enfants.
Conserver à 25 °C (77 °F); excursions permises jusqu'à 15° à 30°C (59° à 86°F) [voir Température ambiante contrôlée USP ].
Le dutastéride est absorbé par la peau. Les gélules d'AVODART 0,5 mg ne doivent pas être manipulées par les femmes enceintes ou susceptibles de le devenir en raison du potentiel d'absorption du dutastéride et du risque potentiel qui en résulte pour un fœtus mâle en développement [voir AVERTISSEMENTS ET PRECAUTIONS ].
Fabriqué pour : GlaxoSmithKline Research Triangle Park. Révisé : janvier 2020
EFFETS SECONDAIRES
Expérience des essais cliniques
Étant donné que les essais cliniques sont menés dans des conditions très variables, les taux d'effets indésirables observés dans les essais cliniques d'un médicament ne peuvent pas être directement comparés aux taux observés dans l'essai clinique d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique.
D'après les essais cliniques avec AVODART 0,5 mg en monothérapie ou en association avec la tamsulosine :
- Les effets indésirables les plus fréquemment rapportés chez les sujets recevant AVODART 0,5 mg étaient l'impuissance, la diminution de la libido, les troubles mammaires (y compris l'hypertrophie et la sensibilité des seins) et les troubles de l'éjaculation. Les effets indésirables les plus fréquemment rapportés chez les sujets recevant un traitement combiné (AVODART 0,5 mg plus tamsulosine) étaient l'impuissance, la diminution de la libido, les troubles mammaires (y compris l'hypertrophie et la sensibilité des seins), les troubles de l'éjaculation et les étourdissements. Les troubles de l'éjaculation sont survenus significativement plus chez les sujets recevant une polythérapie (11 %) que chez ceux recevant AVODART (2 %) ou la tamsulosine (4 %) en monothérapie.
- L'abandon de l'essai en raison d'effets indésirables est survenu chez 4 % des sujets recevant AVODART 0,5 mg et 3 % des sujets recevant le placebo dans les essais contrôlés par placebo avec AVODART. L'effet indésirable le plus fréquent entraînant l'abandon de l'essai était l'impuissance (1 %).
- Dans l'essai clinique évaluant le traitement combiné, l'abandon de l'essai en raison d'effets indésirables est survenu chez 6 % des sujets recevant le traitement combiné (AVODART plus tamsulosine) et 4 % des sujets recevant AVODART 0,5 mg ou la tamsulosine en monothérapie. L'effet indésirable le plus fréquent dans tous les bras de traitement ayant conduit à l'arrêt de l'essai était la dysfonction érectile (1 % à 1,5 %).
Monothérapie
Plus de 4 300 sujets masculins atteints d'HBP ont été répartis au hasard pour recevoir un placebo ou des doses quotidiennes de 0,5 mg d'AVODART dans le cadre de 3 essais de traitement de phase 3 identiques, contrôlés par placebo, en double aveugle, d'une durée de 2 ans, chacun suivi d'une étude ouverte de 2 ans extension. Pendant la période de traitement en double aveugle, 2 167 sujets masculins ont été exposés à AVODART 0,5 mg, dont 1 772 exposés pendant 1 an et 1 510 exposés pendant 2 ans. En incluant les extensions en ouvert, 1 009 sujets masculins ont été exposés à AVODART 0,5 mg pendant 3 ans et 812 ont été exposés pendant 4 ans. La population était âgée de 47 à 94 ans (âge moyen : 66 ans) et plus de 90 % étaient de race blanche. Le tableau 1 résume les effets indésirables cliniques signalés chez au moins 1 % des sujets recevant AVODART et à une incidence plus élevée que les sujets recevant le placebo.
Traitement à long terme (jusqu'à 4 ans)
Cancer de la prostate de haut grade
L'essai REDUCE était un essai randomisé, en double aveugle et contrôlé par placebo qui a recruté 8 231 hommes âgés de 50 à 75 ans avec un PSA sérique de 2,5 ng/mL à 10 ng/mL et une biopsie de la prostate négative au cours des 6 mois précédents. Les sujets ont été randomisés pour recevoir un placebo (n = 4 126) ou des doses quotidiennes de 0,5 mg d'AVODART (n = 4 105) pendant 4 ans maximum. L'âge moyen était de 63 ans et 91 % étaient de race blanche. Les sujets ont subi des biopsies de la prostate programmées conformément au protocole à 2 et 4 ans de traitement ou ont subi des biopsies « pour la cause » à des moments non programmés si cela était cliniquement indiqué. Il y avait une incidence plus élevée de cancer de la prostate avec un score de Gleason de 8 à 10 chez les hommes recevant AVODART (1,0 %) par rapport aux hommes sous placebo (0,5 %) [voir INDICATIONS ET USAGE , AVERTISSEMENTS ET PRECAUTIONS ]. Dans un essai clinique contrôlé par placebo de 7 ans avec un autre inhibiteur de la 5 alpha-réductase (finastéride 5 mg, PROSCAR), des résultats similaires pour le cancer de la prostate avec un score de Gleason de 8 à 10 ont été observés (finastéride 1,8 % contre placebo 1,1 %).
Aucun bénéfice clinique n'a été démontré chez les patients atteints d'un cancer de la prostate traités par AVODART.
Troubles de la reproduction et des seins
Dans les 3 essais pivots contrôlés par placebo sur l'HBP avec AVODART 0,5 mg, d'une durée de 4 ans chacun, il n'y avait aucune preuve d'augmentation des effets indésirables sexuels (impuissance, diminution de la libido et troubles de l'éjaculation) ou de troubles mammaires avec une durée de traitement prolongée. Parmi ces 3 essais, il y a eu 1 cas de cancer du sein dans le groupe dutastéride et 1 cas dans le groupe placebo. Aucun cas de cancer du sein n'a été signalé dans aucun des groupes de traitement de l'essai CombAT de 4 ans ou de l'essai REDUCE de 4 ans.
La relation entre l'utilisation à long terme du dutastéride et la néoplasie du sein chez l'homme est actuellement inconnue.
Combinaison avec la thérapie alpha-bloquante (CombAT)
Plus de 4 800 sujets masculins atteints d'HBP ont été randomisés pour recevoir 0,5 mg d'AVODART 0,5 mg, 0,4 mg de tamsulosine ou une thérapie combinée (0,5 mg d'AVODART plus 0,4 mg de tamsulosine) administrés une fois par jour dans un essai en double aveugle de 4 ans. Au total, 1 623 sujets ont reçu une monothérapie avec AVODART ; 1 611 sujets ont reçu une monothérapie avec la tamsulosine ; et 1 610 sujets ont reçu une polythérapie. La population était âgée de 49 à 88 ans (âge moyen : 66 ans) et 88 % étaient de race blanche. Le tableau 2 résume les effets indésirables rapportés chez au moins 1 % des sujets du groupe association et à une incidence plus élevée que les sujets recevant une monothérapie avec AVODART 0,5 mg ou la tamsulosine.
Insuffisance cardiaque
Dans CombAT, après 4 ans de traitement, l'incidence de l'insuffisance cardiaque à terme composite dans le groupe de thérapie combinée (12/1 610 ; 0,7 %) était plus élevée que dans l'un ou l'autre des groupes de monothérapie : AVODART 0,5 mg, 2/1 623 (0,1 %) et tamsulosine, 9/1 611 (0,6 %). L'insuffisance cardiaque composite a également été examinée dans un essai séparé de 4 ans contrôlé par placebo évaluant AVODART chez les hommes à risque de développer un cancer de la prostate. L'incidence de l'insuffisance cardiaque chez les sujets prenant AVODART 0,5 mg était de 0,6 % (26/4 105) contre 0,4 % (15/4 126) chez les sujets sous placebo. Une majorité de sujets atteints d'insuffisance cardiaque dans les deux essais présentaient des comorbidités associées à un risque accru d'insuffisance cardiaque. Par conséquent, la signification clinique des déséquilibres numériques dans l'insuffisance cardiaque est inconnue. Aucune relation causale entre AVODART seul ou en association avec la tamsulosine et l'insuffisance cardiaque n'a été établie. Aucun déséquilibre n'a été observé dans l'incidence des événements indésirables cardiovasculaires globaux dans les deux essais.
Expérience post-commercialisation
Les effets indésirables suivants ont été identifiés lors de l'utilisation post-approbation d'AVODART. Étant donné que ces réactions sont signalées volontairement par une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale avec l'exposition au médicament. Ces réactions ont été choisies pour être incluses en raison d'une combinaison de leur gravité, de leur fréquence de signalement ou de leur lien de causalité potentiel avec AVODART.
Troubles du système immunitaire
Réactions d'hypersensibilité, y compris éruption cutanée, prurit, urticaire, œdème localisé, réactions cutanées graves et œdème de Quincke.
Tumeurs
Cancer du sein masculin.
Troubles psychiatriques
Humeur dépressive.
Système reproducteur et troubles mammaires
Douleur testiculaire et gonflement testiculaire.
INTERACTIONS MÉDICAMENTEUSES
Inhibiteurs du cytochrome P450 3A
Le dutastéride est largement métabolisé chez l'homme par les isoenzymes du cytochrome P450 (CYP)3A4 et CYP3A5. L'effet des inhibiteurs puissants du CYP3A4 sur le dutastéride n'a pas été étudié. En raison du risque d'interactions médicamenteuses, soyez prudent lorsque vous prescrivez AVODART à des patients prenant de puissants inhibiteurs chroniques de l'enzyme CYP3A4 (par exemple, le ritonavir) [voir PHARMACOLOGIE CLINIQUE ].
Antagonistes alpha-adrénergiques
L'administration d'AVODART en association avec la tamsulosine ou la térazosine n'a aucun effet sur la pharmacocinétique à l'état d'équilibre de l'un ou l'autre des antagonistes alpha-adrénergiques. L'effet de l'administration de tamsulosine ou de térazosine sur les paramètres pharmacocinétiques du dutastéride n'a pas été évalué.
Antagonistes des canaux calciques
L'administration concomitante de vérapamil ou de diltiazem diminue la clairance du dutastéride et entraîne une augmentation de l'exposition au dutastéride. La modification de l'exposition au dutastéride n'est pas considérée comme cliniquement significative. Aucun ajustement posologique n'est recommandé [voir PHARMACOLOGIE CLINIQUE ].
Cholestyramine
L'administration d'une dose unique de 5 mg d'AVODART 0,5 mg suivie 1 heure plus tard de 12 g de cholestyramine n'affecte pas la biodisponibilité relative du dutastéride [voir PHARMACOLOGIE CLINIQUE ].
Digoxine
AVODART 0,5 mg n'altère pas la pharmacocinétique à l'état d'équilibre de la digoxine en cas d'administration concomitante à la dose de 0,5 mg/jour pendant 3 semaines [voir PHARMACOLOGIE CLINIQUE ].
Warfarine
L'administration concomitante d'AVODART 0,5 mg/jour pendant 3 semaines avec de la warfarine ne modifie pas la pharmacocinétique à l'état d'équilibre des isomères S ou R de la warfarine ni n'altère l'effet de la warfarine sur le temps de prothrombine [voir PHARMACOLOGIE CLINIQUE ].
AVERTISSEMENTS
Inclus dans le cadre du PRÉCAUTIONS section.
PRÉCAUTIONS
Effets sur l'antigène spécifique de la prostate (PSA) et l'utilisation du PSA dans la détection du cancer de la prostate
Dans les essais cliniques, AVODART 0,5 mg a réduit la concentration sérique de PSA d'environ 50 % en 3 à 6 mois de traitement. Cette diminution était prévisible sur toute la gamme des valeurs de PSA chez les sujets atteints d'HBP symptomatique, bien qu'elle puisse varier d'un individu à l'autre. AVODART 0,5 mg peut également entraîner une diminution du PSA sérique en présence d'un cancer de la prostate. Pour interpréter les PSA en série chez les hommes prenant AVODART 0,5 mg, une nouvelle ligne de base de PSA doit être établie au moins 3 mois après le début du traitement et le PSA doit être surveillé périodiquement par la suite. Toute augmentation confirmée par rapport à la valeur de PSA la plus basse sous AVODART peut signaler la présence d'un cancer de la prostate et doit être évaluée, même si les taux de PSA se situent toujours dans la plage normale pour les hommes ne prenant pas d'inhibiteur de la 5-alpharéductase. Le non-respect d'AVODART peut également affecter les résultats du test PSA.
Pour interpréter une valeur de PSA isolée chez un homme traité par AVODART 0,5 mg pendant 3 mois ou plus, la valeur de PSA doit être doublée pour comparaison avec les valeurs normales chez les hommes non traités. Le rapport PSA libre sur total (pourcentage de PSA libre) reste constant, même sous l'influence d'AVODART. Si les cliniciens choisissent d'utiliser le pourcentage de PSA libre comme aide à la détection du cancer de la prostate chez les hommes recevant AVODART, aucun ajustement de sa valeur ne semble nécessaire.
L'administration concomitante de dutastéride et de tamsulosine a entraîné des modifications du PSA sérique similaires à celles du dutastéride en monothérapie.
Risque accru de cancer de la prostate de haut grade
Chez les hommes âgés de 50 à 75 ans avec une biopsie antérieure négative pour le cancer de la prostate et un PSA de base compris entre 2,5 ng/mL et 10,0 ng/mL prenant AVODART dans l'essai de 4 ans REDUCE (Reduction by Dutasteride of Prostate Cancer Events), il y avait une augmentation de l'incidence du cancer de la prostate de score de Gleason 810 par rapport aux hommes prenant un placebo (AVODART 1,0 % versus placebo 0,5 %) [voir INDICATIONS ET USAGE , EFFETS INDÉSIRABLES ]. Dans un essai clinique contrôlé par placebo de 7 ans avec un autre inhibiteur de la 5 alpha-réductase (finastéride 5 mg, PROSCAR), des résultats similaires pour le cancer de la prostate avec un score de Gleason de 8 à 10 ont été observés (finastéride 1,8 % contre placebo 1,1 %).
Les inhibiteurs de la 5 alpha-réductase peuvent augmenter le risque de développer un cancer de la prostate de haut grade. Il n'a pas été établi si l'effet des inhibiteurs de la 5 alpha-réductase sur la réduction du volume de la prostate ou des facteurs liés aux essais ont eu un impact sur les résultats de ces essais.
Évaluation pour d'autres maladies urologiques
Avant d'initier un traitement par AVODART 0,5 mg, il convient de tenir compte d'autres affections urologiques susceptibles de provoquer des symptômes similaires. De plus, l'HBP et le cancer de la prostate peuvent coexister.
Exposition transdermique à AVODART 0,5 mg chez les femmes enceintes - risque pour le fœtus masculin
Les gélules AVODART ne doivent pas être manipulées par les femmes enceintes ou susceptibles de l'être. Le dutastéride peut être absorbé par la peau et pourrait entraîner une exposition fœtale involontaire et un risque potentiel pour un fœtus de sexe masculin. Si une femme enceinte entre en contact avec des gélules de dutastéride qui fuient, la zone de contact doit être immédiatement lavée à l'eau et au savon [voir Utilisation dans des populations spécifiques ]. Le dutastéride peut être absorbé par la peau selon des études sur des animaux [voir Toxicologie non clinique ].
Don de sang
Les hommes traités par AVODART ne doivent pas donner de sang avant qu'au moins 6 mois ne se soient écoulés après leur dernière dose. Le but de cette période différée est d'empêcher l'administration de dutastéride à une femme enceinte transfusée.
Effet sur les caractéristiques du sperme
Les effets du dutastéride 0,5 mg/jour sur les caractéristiques du sperme ont été évalués chez des hommes sains pendant 52 semaines de traitement et 24 semaines de suivi post-traitement. À 52 semaines, par rapport au placebo, le traitement par dutastéride a entraîné une réduction moyenne du nombre total de spermatozoïdes, du volume de sperme et de la motilité des spermatozoïdes ; les effets sur le nombre total de spermatozoïdes n'étaient pas réversibles après 24 semaines de suivi. La concentration et la morphologie des spermatozoïdes n'ont pas été affectées et les valeurs moyennes de tous les paramètres du sperme sont restées dans la plage normale à tous les moments. La signification clinique de l'effet du dutastéride sur les caractéristiques du sperme pour la fertilité d'un patient individuel n'est pas connue [voir Utilisation dans des populations spécifiques ].
Informations sur les conseils aux patients
Conseillez au patient de lire l'étiquetage patient approuvé par la FDA ( INFORMATIONS PATIENTS ).
Surveillance de l'APS
Informez les patients qu'AVODART réduit les taux sériques de PSA d'environ 50 % dans les 3 à 6 mois suivant le traitement, bien que cela puisse varier d'un individu à l'autre. Pour les patients subissant un dépistage de l'APS, une augmentation des taux d'APS pendant le traitement par AVODART peut signaler la présence d'un cancer de la prostate et doit être évaluée par un professionnel de la santé [voir AVERTISSEMENTS ET PRECAUTIONS ].
Risque accru de cancer de la prostate de haut grade
Informer les patients qu'il y a eu une augmentation du cancer de la prostate de haut grade chez les hommes traités avec des inhibiteurs de la 5 alpha-réductase (qui sont indiqués pour le traitement de l'HBP), y compris AVODART 0,5 mg, par rapport à ceux traités avec un placebo dans les essais portant sur l'utilisation de ces médicaments pour réduire le risque de cancer de la prostate [voir INDICATIONS ET USAGE , AVERTISSEMENTS ET PRECAUTIONS , EFFETS INDÉSIRABLES ].
Exposition transdermique d'AVODART chez les femmes enceintes ou potentiellement enceintes - risque pour le fœtus masculin
Informez les patients qu'AVODART 0,5 mg gélules ne doit pas être manipulé par les femmes enceintes ou susceptibles de l'être en raison du potentiel d'absorption du dutastéride et du risque potentiel qui en résulte pour un fœtus masculin en développement. Le dutastéride peut être absorbé par la peau et pourrait entraîner une exposition fœtale accidentelle. Si une femme enceinte ou potentiellement enceinte entre en contact avec des gélules d'AVODART 0,5 mg qui fuient, la zone de contact doit être immédiatement lavée à l'eau et au savon [voir AVERTISSEMENTS ET PRECAUTIONS , Utilisation dans des populations spécifiques ].
Effets sur les paramètres du sperme
Informer les hommes qu'AVODART 0,5 mg peut affecter les caractéristiques du sperme mais que l'effet sur la fertilité est inconnu [voir AVERTISSEMENTS ET PRECAUTIONS , Utilisation dans des populations spécifiques ].
Don de sang
Informez les hommes traités par AVODART 0,5 mg qu'ils ne doivent pas donner de sang avant au moins 6 mois après leur dernière dose afin d'éviter que les femmes enceintes ne reçoivent du dutastéride par transfusion sanguine [voir AVERTISSEMENTS ET PRECAUTIONS ]. Les taux sériques de dutastéride sont détectables pendant 4 à 6 mois après la fin du traitement [voir PHARMACOLOGIE CLINIQUE ].
AVODART est une marque déposée détenue ou sous licence par le groupe de sociétés GSK.
Les autres marques répertoriées sont des marques commerciales détenues ou concédées sous licence à leurs propriétaires respectifs et ne sont pas détenues ou concédées sous licence au groupe de sociétés GSK. Les fabricants de ces marques ne sont pas affiliés et ne cautionnent pas le groupe de sociétés GSK ou ses produits.
Toxicologie non clinique
Carcinogenèse, mutagenèse, altération de la fertilité
Carcinogenèse
Une étude de cancérogénicité de 2 ans a été menée chez des souris B6C3F1 à des doses de 3, 35, 250 et 500 mg/kg/jour pour les mâles et de 3, 35 et 250 mg/kg/jour pour les femelles ; une incidence accrue d'adénomes hépatocellulaires bénins a été notée à 250 mg/kg/jour (290 fois la MRHD d'une dose quotidienne de 0,5 mg) chez les souris femelles uniquement. Deux des 3 principaux métabolites humains ont été détectés chez la souris. L'exposition à ces métabolites chez la souris est soit plus faible que chez l'homme, soit inconnue.
Dans une étude de cancérogénicité de 2 ans chez des rats Han Wistar, à des doses de 1,5, 7,5 et 53 mg/kg/jour chez les mâles et de 0,8, 6,3 et 15 mg/kg/jour chez les femelles, il y a eu une augmentation de la cellule de Leydig adénomes dans les testicules à 135 fois la MRHD (53 mg/kg/jour et plus). Une incidence accrue d'hyperplasie des cellules de Leydig était présente à 52 fois la MRHD (doses de rats mâles de 7,5 mg/kg/jour et plus). Une corrélation positive entre les changements prolifératifs dans les cellules de Leydig et une augmentation des taux circulants d'hormone lutéinisante a été démontrée avec les inhibiteurs de la 5 alpha-réductase et est compatible avec un effet sur l'axe hypothalamo-hypophyso-testiculaire suite à l'inhibition de la 5 alpha-réductase. Aux doses tumorigènes, les taux d'hormone lutéinisante chez le rat ont augmenté de 167 %. Dans cette étude, les principaux métabolites humains ont été testés pour la cancérogénicité à environ 1 à 3 fois l'exposition clinique attendue.
Mutagenèse
Le dutastéride a été testé pour sa génotoxicité dans un test de mutagenèse bactérienne (test d'Ames), un test d'aberration chromosomique sur des cellules ovariennes de hamster chinois et un test du micronoyau chez le rat. Les résultats n'ont pas indiqué de potentiel génotoxique de la molécule mère. Deux principaux métabolites humains étaient également négatifs soit au test d'Ames, soit à un test d'Ames abrégé.
Altération de la fertilité
Le traitement de rats mâles sexuellement matures avec du dutastéride à 0,1 fois la MRHD (doses animales de 0,05 mg/kg/jour ou plus pendant jusqu'à 31 semaines) sur la base de la concentration sérique moyenne a entraîné une diminution dose-dépendante du temps de la fertilité à toutes les doses ; réduction du nombre (absolu) de spermatozoïdes de la caudale épididymaire, mais pas de la concentration des spermatozoïdes (à 50 et 500 mg/kg/jour); réduction du poids de l'épididyme, de la prostate et des vésicules séminales ; et modifications microscopiques (vacuolisation cytoplasmique de l'épithélium tubulaire dans les épididymes et/ou diminution du contenu cytoplasmique de l'épithélium, compatible avec une diminution de l'activité sécrétoire dans la prostate et les vésicules séminales) dans les organes reproducteurs à toutes les doses en l'absence de toxicité paternelle. Les effets sur la fertilité ont été inversés à la semaine de récupération 6 à toutes les doses, et le nombre de spermatozoïdes était normal à la fin d'une période de récupération de 14 semaines. Les changements microscopiques n'étaient plus présents à la semaine de récupération 14 à 0,1 fois la MRHD et ont été partiellement récupérés dans les groupes de traitement restants. De faibles niveaux de dutastéride (0,6 à 17 ng/mL) ont été détectés dans le sérum de rats femelles non traités accouplés à des mâles traités (10 à 500 mg/kg/jour pendant 29 à 30 semaines) qui sont de 16 à 110 fois la MRHD basée sur concentration sérique moyenne. Aucune féminisation n'est survenue chez les descendants mâles de rats femelles non traités accouplés à des rats mâles traités, même si des taux sanguins détectables de dutastéride ont été observés chez les rats femelles.
Dans une étude de fertilité chez des rats femelles avec une administration 4 semaines avant l'accouplement jusqu'au début de la gestation, l'administration orale de dutastéride à des doses de 0,05, 2,5, 12,5 et 30 mg/kg/jour a entraîné une réduction de la taille de la portée en raison de l'augmentation des résorptions et de la féminisation de fœtus mâles (diminution de la distance anogénitale) à 2 à 10 fois la MRHD (doses animales de 2,5 mg/kg/jour ou plus) sur la base de la concentration sérique moyenne, en présence de toxicité maternelle (diminution du gain de poids corporel). Les poids corporels fœtaux ont également été réduits à environ 0,02 fois la MRHD (dose chez le rat de 0,05 mg/kg/jour ou plus) sur la base de la concentration sérique moyenne, sans niveau sans effet, en l'absence de toxicité maternelle.
Utilisation dans des populations spécifiques
Grossesse
Résumé des risques
AVODART 0,5 mg est contre-indiqué pendant la grossesse car il peut être nocif pour le fœtus mâle [voir CONTRE-INDICATIONS ]. AVODART n'est pas indiqué chez la femme.
AVODART 0,5 mg est un inhibiteur de la 5 alpha-réductase qui empêche la conversion de la testostérone en dihydrotestostérone (DHT), une hormone nécessaire au développement normal des organes génitaux masculins. Les anomalies des organes génitaux des fœtus mâles sont une conséquence physiologique attendue de l'inhibition de cette conversion. Ces résultats sont similaires aux observations chez les nourrissons de sexe masculin présentant un déficit génétique en 5 alpha-réductase.
Dans la population générale des États-Unis, le risque de fond estimé de malformations congénitales majeures et de fausse couche dans les grossesses cliniquement reconnues est de 2 % à 4 % et de 15 % à 20 %, respectivement.
Dans les études de reproduction chez l'animal, le dutastéride a inhibé le développement normal des organes génitaux externes chez la progéniture mâle lorsqu'il a été administré à des rats ou des lapins pendant l'organogenèse à une dose inférieure à la dose humaine maximale recommandée (MRHD) de 0,5 mg par jour, en l'absence de toxicité maternelle. À 15 fois la MRHD, une grossesse prolongée, une diminution du poids des organes reproducteurs et une puberté retardée chez la progéniture mâle ont été observées chez les rats, avec des niveaux sans effet inférieurs à la MRHD de 0,5 mg par jour. Une augmentation du poids placentaire chez les lapins a également été observée, avec des doses sans effet inférieures à la MRHD de 0,5 mg par jour (voir Données ).
Bien que le dutastéride soit sécrété dans le sperme humain, la concentration du médicament chez la partenaire féminine humaine est environ 100 fois inférieure aux concentrations produisant des anomalies des organes génitaux masculins dans les études animales (voir Données ). Chez les singes ayant reçu au cours de l'organogenèse des concentrations sanguines comparables ou supérieures aux niveaux auxquels on estime qu'une partenaire féminine humaine est exposée, les organes génitaux externes de la progéniture mâle n'ont pas été affectés. Aucune féminisation n'est survenue chez les descendants mâles de rats femelles non traités accouplés à des rats mâles traités, même si des taux sanguins détectables de dutastéride ont été observés chez les rats femelles [voir Toxicologie non clinique ].
Données
Données humaines
La concentration la plus élevée mesurée de dutastéride dans le sperme chez les hommes traités était de 14 ng/mL. Bien que le dutastéride soit détecté dans le sperme, en supposant l'exposition d'une femme de 50 kg à 5 mL de sperme et une absorption de 100 %, la concentration sanguine attendue de dutastéride dans le sperme serait d'environ 0,0175 ng/mL. Cette concentration est environ 100 fois inférieure aux concentrations sanguines produisant des anomalies des organes génitaux mâles dans les études animales. Le dutastéride est fortement lié aux protéines dans le sperme humain (plus de 96 %), ce qui peut réduire la quantité de dutastéride disponible pour l'absorption vaginale.
Données animales
Dans une étude sur le développement embryo-fœtal chez le rat, l'administration orale de dutastéride à 10 fois moins que la MRHD de 0,5 mg par jour (sur la base des taux sanguins moyens chez l'homme) a entraîné une féminisation des organes génitaux masculins chez le fœtus (diminution de la distance anogénitale à 0,05 mg /kg/jour, en l'absence de dose sans effet) en l'absence de toxicité maternelle. De plus, le développement du mamelon, l'hypospadias et les glandes préputiales distendues se sont produits chez les fœtus de mères traitées à des doses de 2,5 mg/kg/jour ou plus (environ 15 fois la MRHD). Une réduction du poids corporel fœtal et une ossification retardée associée en présence de toxicité maternelle (diminution du gain de poids corporel) ont été observées à une exposition maternelle d'environ 15 fois la MRHD (dose de 2,5 mg/kg/jour ou plus). Une augmentation des ratons mort-nés a été observée chez les mères traitées à 30 mg/kg/jour (environ 111 fois la MRHD), avec un niveau sans effet de 12,5 mg/kg/jour.
Dans une étude sur le développement embryo-fœtal chez le lapin, des doses 28 fois la MRHD (doses de 30 mg/kg/jour ou plus), basées sur les concentrations sanguines moyennes chez l'homme, ont été administrées par voie orale les jours de gestation 7 à 29 (pendant l'organogenèse et la fin période de développement des organes génitaux externes). L'évaluation histologique de la papille génitale des fœtus a révélé des signes de féminisation du fœtus mâle ainsi que des os du crâne fusionnés et une augmentation du poids du placenta à toutes les doses en l'absence de toxicité maternelle. Une deuxième étude sur le développement embryo-fœtal chez des lapins dosés tout au long de la grossesse (organogenèse et période ultérieure de développement des organes génitaux externes [jours de gestation 6 à 29]) à 0,3 fois la MRHD (doses de 0,05 mg/kg/jour ou plus, sans aucune niveau d'effet), a également produit des preuves de féminisation des organes génitaux chez les fœtus mâles et d'augmentation du poids du placenta à toutes les doses en l'absence de toxicité maternelle.
Dans une étude sur le développement embryo-fœtal, des singes rhésus gravides ont été exposés par voie intraveineuse pendant l'organogenèse (jours de gestation 20 à 100) à un taux sanguin de dutastéride comparable ou supérieur à l'exposition estimée au dutastéride d'une partenaire humaine. Le dutastéride a été administré du 20e au 100e jour de gestation (pendant l'organogenèse) à des doses de 400, 780, 1 325 ou 2 010 ng/jour (12 singes/groupe). Aucune féminisation des organes génitaux externes mâles de la progéniture de singe n'a été observée. Une réduction du poids des surrénales fœtales, une réduction du poids de la prostate fœtale et une augmentation du poids des ovaires et des testicules fœtaux ont été observées à la dose la plus élevée testée. Sur la base de la plus forte concentration mesurée de sperme de dutastéride chez les hommes traités (14 ng/mL), ces doses chez le singe représentent jusqu'à 16 fois l'exposition maximale potentielle d'une femme humaine de 50 kg à 5 ml de sperme par jour provenant d'un dutastéride- mâle traité, en supposant une absorption de 100 %. Les niveaux de dose (en ng/kg) administrés aux singes dans cette étude sont de 32 à 186 fois la dose nominale (ng/kg) à laquelle une femelle serait potentiellement exposée par le sperme. On ne sait pas si les lapins ou les singes rhésus produisent l'un des principaux métabolites humains.
Dans une étude sur le développement oral prénatal et postnatal chez le rat, une féminisation des organes génitaux masculins a été observée. Une diminution de la distance anogénitale a été observée à 0,05 fois la MRHD et plus (0,05 mg/kg/jour et plus), avec l'absence d'un niveau sans effet, basé sur les taux sanguins moyens chez les hommes comme estimation de l'ASC. Des hypospadias et le développement des mamelons ont été observés à 2,5 mg/kg/jour ou plus (14 fois la MRHD ou plus, avec un niveau sans effet à 0,05 mg/kg/jour). Des doses de 2,5 mg/kg/jour et plus ont également entraîné une prolongation de la gestation chez les femelles parentales, une augmentation du temps jusqu'à la séparation balano-préputiale chez les descendants mâles, une diminution du temps jusqu'à la perméabilité vaginale chez les descendants femelles et une diminution de la prostate et poids des vésicules séminales chez les descendants mâles. Une augmentation des mortinaissances et une diminution de la viabilité néonatale chez la progéniture ont été notées à 30 mg/kg/jour (102 fois la MRHD en présence de toxicité maternelle [diminution du poids corporel]).
Lactation
Résumé des risques
AVODART n'est pas indiqué chez la femme. Aucune information n'est disponible sur la présence de dutastéride dans le lait maternel, les effets sur l'enfant allaité ou les effets sur la production de lait.
Femelles et mâles de potentiel reproducteur
Infertilité
Mâles
Les effets du dutastéride 0,5 mg/jour sur les caractéristiques du sperme ont été évalués chez des volontaires sains âgés de 18 à 52 ans (n = 27 dutastéride, n = 23 placebo) pendant 52 semaines de traitement et 24 semaines de suivi post-traitement. À 52 semaines, les réductions moyennes en pourcentage par rapport au départ du nombre total de spermatozoïdes, du volume de sperme et de la motilité des spermatozoïdes étaient de 23 %, 26 % et 18 %, respectivement, dans le groupe dutastéride après ajustement des changements par rapport au départ dans le groupe placebo. La concentration et la morphologie des spermatozoïdes n'ont pas été affectées. Après 24 semaines de suivi, la variation moyenne en pourcentage du nombre total de spermatozoïdes dans le groupe dutastéride est restée inférieure de 23 % à la valeur initiale. Alors que les valeurs moyennes de tous les paramètres du sperme à tous les moments sont restées dans les plages normales et n'ont pas répondu aux critères prédéfinis d'un changement cliniquement significatif (30 %), 2 sujets du groupe dutastéride ont présenté une diminution du nombre de spermatozoïdes supérieure à 90 % par rapport au départ à 52 semaines, avec récupération partielle au suivi de 24 semaines. La signification clinique de l'effet du dutastéride sur les caractéristiques du sperme pour la fertilité d'un patient individuel n'est pas connue [voir AVERTISSEMENTS ET PRECAUTIONS ].
Utilisation pédiatrique
AVODART 0,5 mg n'est pas indiqué chez les patients pédiatriques. L'innocuité et l'efficacité chez les patients pédiatriques n'ont pas été établies.
Utilisation gériatrique
Sur 2 167 sujets masculins traités par AVODART 0,5 mg dans 3 essais cliniques, 60 % étaient âgés de 65 ans et plus et 15 % étaient âgés de 75 ans et plus. Aucune différence globale d'innocuité ou d'efficacité n'a été observée entre ces sujets et les sujets plus jeunes. D'autres expériences cliniques rapportées n'ont pas identifié de différences dans les réponses entre les patients âgés et les patients plus jeunes, mais une plus grande sensibilité de certains individus plus âgés ne peut être exclue [voir PHARMACOLOGIE CLINIQUE ].
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire pour AVODART 0,5 mg chez les patients insuffisants rénaux [voir PHARMACOLOGIE CLINIQUE ].
Insuffisance hépatique
L'effet de l'insuffisance hépatique sur la pharmacocinétique du dutastéride n'a pas été étudié. Le dutastéride étant largement métabolisé, l'exposition pourrait être plus élevée chez les patients insuffisants hépatiques. Cependant, dans un essai clinique où 60 sujets ont reçu 5 mg (10 fois la dose thérapeutique) par jour pendant 24 semaines, aucun événement indésirable supplémentaire n'a été observé par rapport à ceux observés à la dose thérapeutique de 0,5 mg [voir PHARMACOLOGIE CLINIQUE ].
SURDOSAGE
Dans des essais volontaires, des doses uniques de dutastéride allant jusqu'à 40 mg (80 fois la dose thérapeutique) pendant 7 jours ont été administrées sans problèmes de sécurité significatifs. Dans un essai clinique, des doses quotidiennes de 5 mg (10 fois la dose thérapeutique) ont été administrées à 60 sujets pendant 6 mois sans effets indésirables supplémentaires à ceux observés à des doses thérapeutiques de 0,5 mg.
Il n'existe pas d'antidote spécifique au dutastéride. Par conséquent, en cas de suspicion de surdosage, un traitement symptomatique et de soutien doit être administré selon les besoins, en tenant compte de la longue demi-vie du dutastéride.
CONTRE-INDICATIONS
AVODART 0,5 mg est contre-indiqué chez :

- Grossesse. L'utilisation du dutastéride est contre-indiquée chez les femmes enceintes. Dans les études de toxicité pour la reproduction et le développement chez l'animal, le dutastéride a inhibé le développement des organes génitaux externes du fœtus mâle. Par conséquent, AVODART 0,5 mg peut être nocif pour le fœtus lorsqu'il est administré à une femme enceinte [voir AVERTISSEMENTS ET PRECAUTIONS , Utilisation dans des populations spécifiques ].
- Patients ayant précédemment démontré une hypersensibilité cliniquement significative (p. ex., réactions cutanées graves, œdème de Quincke) à AVODART ou à d'autres inhibiteurs de la 5 alpha-réductase [voir EFFETS INDÉSIRABLES ].
PHARMACOLOGIE CLINIQUE
Mécanisme d'action
Le dutastéride inhibe la conversion de la testostérone en DHT. La DHT est l'androgène principalement responsable du développement initial et de l'élargissement ultérieur de la prostate. La testostérone est convertie en DHT par l'enzyme 5 alpha-réductase, qui existe sous 2 isoformes, type 1 et type 2. L'isoenzyme de type 2 est principalement active dans les tissus reproducteurs, tandis que l'isoenzyme de type 1 est également responsable de la conversion de la testostérone dans le peau et foie.
Le dutastéride est un inhibiteur compétitif et spécifique des isoenzymes 5 alpha-réductases de type 1 et de type 2, avec lesquelles il forme un complexe enzymatique stable. La dissociation de ce complexe a été évaluée dans des conditions in vitro et in vivo et est extrêmement lente. Le dutastéride ne se lie pas au récepteur humain des androgènes.
Pharmacodynamie
Effet sur la 5 alpha-dihydrotestostérone et la testostérone
L'effet maximal des doses quotidiennes de dutastéride sur la réduction de la DHT est dose-dépendant et est observé en 1 à 2 semaines. Après 1 et 2 semaines d'administration quotidienne de dutastéride 0,5 mg, les concentrations sériques médianes de DHT ont été réduites de 85 % et 90 %, respectivement. Chez les patients atteints d'HBP traités par dutastéride 0,5 mg/jour pendant 4 ans, la diminution médiane de la DHT sérique était de 94 % à 1 an, de 93 % à 2 ans et de 95 % à 3 et 4 ans. L'augmentation médiane de la testostérone sérique était de 19 % à 1 et 2 ans, de 26 % à 3 ans et de 22 % à 4 ans, mais les niveaux moyen et médian sont restés dans les limites physiologiques.
Chez les patients atteints d'HBP traités avec 5 mg/jour de dutastéride ou un placebo jusqu'à 12 semaines avant la résection transurétrale de la prostate, les concentrations moyennes de DHT dans le tissu prostatique étaient significativement plus faibles dans le groupe dutastéride par rapport au placebo (784 et 5 793 pg/g , respectivement, P
Les hommes adultes atteints d'un déficit génétiquement hérité de type 2 5 alpha-réductase ont également des taux de DHT diminués. Ces hommes déficients en 5 alpha-réductase ont une petite prostate tout au long de leur vie et ne développent pas d'HBP. À l'exception des malformations urogénitales associées présentes à la naissance, aucune autre anomalie clinique liée à un déficit en 5 alpha-réductase n'a été observée chez ces personnes.
Effets sur d'autres hormones
Chez des volontaires sains, 52 semaines de traitement par dutastéride 0,5 mg/jour (n = 26) n'ont entraîné aucun changement cliniquement significatif par rapport au placebo (n = 23) en globuline liant les hormones sexuelles, estradiol, hormone lutéinisante, hormone folliculo-stimulante, la thyroxine (T4 libre) et la déhydroépiandrostérone. Des augmentations moyennes statistiquement significatives et ajustées au départ par rapport au placebo ont été observées pour la testostérone totale à 8 semaines (97,1 ng/dL, P
Autres effets
Le bilan lipidique plasmatique et la densité minérale osseuse ont été évalués après 52 semaines de dutastéride 0,5 mg une fois par jour chez des volontaires sains. Il n'y a eu aucun changement dans la densité minérale osseuse mesurée par absorptiométrie à rayons X à double énergie par rapport au placebo ou à la ligne de base. De plus, le profil lipidique plasmatique (c.-à-d. cholestérol total, lipoprotéines de faible densité, lipoprotéines de haute densité, triglycérides) n'a pas été modifié par le dutastéride. Aucun changement cliniquement significatif dans les réponses hormonales surrénales à la stimulation de l'hormone corticotrope (ACTH) n'a été observé dans un sous-ensemble de population (n = 13) de l'essai de volontaires sains d'un an.
Pharmacocinétique
Absorption
Après l'administration d'une dose unique de 0,5 mg d'une capsule de gélatine molle, le temps nécessaire pour atteindre les concentrations sériques maximales (Tmax) du dutastéride est de 2 à 3 heures. La biodisponibilité absolue chez 5 sujets sains est d'environ 60 % (intervalle : 40 % à 94 %). Lorsque le médicament est administré avec de la nourriture, les concentrations sériques maximales ont été réduites de 10 à 15 %. Cette réduction n'a aucune signification clinique.
Distribution
Les données pharmacocinétiques après administration orale unique et répétée montrent que le dutastéride a un grand volume de distribution (300 à 500 L). Le dutastéride est fortement lié à l'albumine plasmatique (99,0 %) et à la glycoprotéine acide alpha-1 (96,6 %).
Dans un essai portant sur des sujets sains (n = 26) recevant du dutastéride à raison de 0,5 mg/jour pendant 12 mois, les concentrations de dutastéride dans le sperme étaient en moyenne de 3,4 ng/mL (intervalle : 0,4 à 14 ng/mL) à 12 mois et, comme dans le sérum, ont atteint une stabilité -état des concentrations à 6 mois. En moyenne, à 12 mois, 11,5 % des concentrations sériques de dutastéride se sont réparties dans le sperme.
Métabolisme et élimination
Le dutastéride est largement métabolisé chez l'homme. Des études in vitro ont montré que le dutastéride est métabolisé par les isoenzymes CYP3A4 et CYP3A5. Ces deux isoenzymes ont produit les métabolites 4'-hydroxydutastéride, 6-hydroxydutastéride et 6,4'-dihydroxydutastéride. De plus, le métabolite 15-hydroxydutastéride a été formé par le CYP3A4. Le dutastéride n'est pas métabolisé in vitro par les isoenzymes du cytochrome P450 humain CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 et CYP2E1. Dans le sérum humain après administration à l'état d'équilibre, dutastéride inchangé, 3 métabolites majeurs (4'-hydroxydutastéride, 1,2-dihydrodutastéride et 6-hydroxydutastéride) et 2 métabolites mineurs (6,4'-dihydroxydutastéride et 15 -hydroxydutastéride), évalués par la réponse par spectrométrie de masse, ont été détectés. La stéréochimie absolue des additions d'hydroxyle dans les positions 6 et 15 n'est pas connue. In vitro, les métabolites 4'-hydroxydutastéride et 1,2-dihydrodutastéride sont beaucoup moins puissants que le dutastéride contre les deux isoformes de la 5 alpha-réductase humaine. L'activité du 6β-hydroxydutastéride est comparable à celle du dutastéride.
Le dutastéride et ses métabolites ont été principalement excrétés dans les fèces. En pourcentage de la dose, il y avait environ 5 % de dutastéride inchangé (~1 % à ~15 %) et 40 % sous forme de métabolites apparentés au dutastéride (~2 % à ~90 %). Seules des traces de dutastéride inchangé ont été retrouvées dans les urines (
La demi-vie d'élimination terminale du dutastéride est d'environ 5 semaines à l'état d'équilibre. La concentration sérique moyenne de dutastéride à l'état d'équilibre était de 40 ng/mL après 0,5 mg/jour pendant 1 an. Après administration quotidienne, les concentrations sériques de dutastéride atteignent 65 % de la concentration à l'état d'équilibre après 1 mois et environ 90 % après 3 mois. En raison de la longue demi-vie du dutastéride, les concentrations sériques restent détectables (supérieures à 0,1 ng/mL) jusqu'à 4 à 6 mois après l'arrêt du traitement.
Populations spécifiques
Patients pédiatriques
La pharmacocinétique du dutastéride n'a pas été étudiée chez les sujets de moins de 18 ans.
Patients gériatriques
Aucun ajustement posologique n'est nécessaire chez les personnes âgées. La pharmacocinétique et la pharmacodynamique du dutastéride ont été évaluées chez 36 sujets sains de sexe masculin âgés de 24 à 87 ans après administration d'une dose unique de 5 mg de dutastéride. Dans cet essai à dose unique, la demi-vie du dutastéride a augmenté avec l'âge (environ 170 heures chez les hommes de 20 à 49 ans, environ 260 heures chez les hommes de 50 à 69 ans et environ 300 heures chez les hommes de plus de 70 ans). Sur 2 167 hommes traités par dutastéride dans les 3 essais pivots, 60 % étaient âgés de 65 ans et plus et 15 % avaient 75 ans et plus. Aucune différence globale d'innocuité ou d'efficacité n'a été observée entre ces patients et les patients plus jeunes.
Patients masculins et féminins
AVODART est contre-indiqué pendant la grossesse et n'est pas indiqué chez la femme [voir CONTRE-INDICATIONS , AVERTISSEMENTS ET PRECAUTIONS ]. La pharmacocinétique du dutastéride chez la femme n'a pas été étudiée.
Groupes raciaux et ethniques
L'effet de la race sur la pharmacocinétique du dutastéride n'a pas été étudié.
Patients atteints d'insuffisance rénale
L'effet de l'insuffisance rénale sur la pharmacocinétique du dutastéride n'a pas été étudié. Cependant, moins de 0,1 % d'une dose de 0,5 mg de dutastéride à l'état d'équilibre est récupérée dans l'urine humaine, de sorte qu'aucun ajustement de la posologie n'est prévu pour les patients atteints d'insuffisance rénale.
Patients atteints d'insuffisance hépatique
L'effet de l'insuffisance hépatique sur la pharmacocinétique du dutastéride n'a pas été étudié. Le dutastéride étant largement métabolisé, l'exposition pourrait être plus élevée chez les patients insuffisants hépatiques.
Études sur les interactions médicamenteuses
Inhibiteurs du cytochrome P450
Aucun essai clinique d'interaction médicamenteuse n'a été réalisé pour évaluer l'impact des inhibiteurs de l'enzyme CYP3A sur la pharmacocinétique du dutastéride. Cependant, sur la base de données in vitro, les concentrations sanguines de dutastéride peuvent augmenter en présence d'inhibiteurs du CYP3A4/5 tels que le ritonavir, le kétoconazole, le vérapamil, le diltiazem, la cimétidine, la troléandomycine et la ciprofloxacine.
Le dutastéride n'inhibe pas le métabolisme in vitro des substrats modèles des principales isoenzymes du cytochrome P450 humain (CYP1A2, CYP2C9, CYP2C19, CYP2D6 et CYP3A4) à une concentration de 1 000 ng/mL, 25 fois supérieure aux concentrations sériques à l'état d'équilibre chez l'homme .
Antagonistes alpha-adrénergiques
Dans un essai croisé à séquence unique chez des volontaires sains, l'administration de tamsulosine ou de térazosine en association avec AVODART n'a eu aucun effet sur la pharmacocinétique à l'état d'équilibre de l'un ou l'autre des antagonistes alpha-adrénergiques. Bien que l'effet de l'administration de tamsulosine ou de térazosine sur les paramètres pharmacocinétiques du dutastéride n'ait pas été évalué, le pourcentage de variation des concentrations de DHT était similaire pour AVODART 0,5 mg seul par rapport au traitement combiné.
Antagonistes des canaux calciques
Dans une analyse pharmacocinétique de population, une diminution de la clairance du dutastéride a été notée lorsqu'il était co-administré avec les inhibiteurs du CYP3A4, le vérapamil (-37 %, n = 6) et le diltiazem (-44 %, n = 5). En revanche, aucune diminution de la clairance n'a été observée lorsque l'amlodipine, un autre antagoniste des canaux calciques qui n'est pas un inhibiteur du CYP3A4, a été co-administrée avec le dutastéride (+7 %, n = 4).
La diminution de la clairance et l'augmentation subséquente de l'exposition au dutastéride en présence de vérapamil et de diltiazem ne sont pas considérées comme cliniquement significatives. Aucun ajustement posologique n'est recommandé.
Cholestyramine
L'administration d'une dose unique de 5 mg d'AVODART 0,5 mg suivie 1 heure plus tard de 12 g de cholestyramine n'a pas affecté la biodisponibilité relative du dutastéride chez 12 volontaires sains.
Digoxine
Dans un essai portant sur 20 volontaires sains, AVODART 0,5 mg n'a pas altéré la pharmacocinétique à l'état d'équilibre de la digoxine lorsqu'il est administré en concomitance à une dose de 0,5 mg/jour pendant 3 semaines.
Warfarine
Dans un essai mené auprès de 23 volontaires sains, 3 semaines de traitement par AVODART 0,5 mg/jour n'ont pas modifié la pharmacocinétique à l'état d'équilibre des isomères S ou R de la warfarine ni modifié l'effet de la warfarine sur le temps de prothrombine lorsqu'elle est administrée avec la warfarine.
Autre thérapie concomitante
Bien qu'aucun essai d'interaction spécifique n'ait été réalisé avec d'autres composés, environ 90 % des sujets des 3 essais randomisés, en double aveugle, contrôlés par placebo sur l'innocuité et l'efficacité recevant AVODART prenaient d'autres médicaments en concomitance. Aucune interaction indésirable cliniquement significative n'a pu être attribuée à l'association d'AVODART et d'un traitement concomitant lorsqu'AVODART était co-administré avec des anti-hyperlipidémiques, des inhibiteurs de l'enzyme de conversion de l'angiotensine (ECA), des agents bêta-bloquants, des inhibiteurs calciques, des corticostéroïdes, des diurétiques, des anti-inflammatoires non stéroïdiens. - les médicaments inflammatoires (AINS), les inhibiteurs de la phosphodiestérase de type V et les antibiotiques quinolones.
Toxicologie animale et/ou pharmacologie
Études de toxicologie du système nerveux central
Chez les rats et les chiens, l'administration orale répétée de dutastéride a entraîné chez certains animaux des signes de toxicité non spécifique, réversible et à médiation centrale sans modifications histopathologiques associées à des expositions 425 et 315 fois l'exposition clinique attendue (du médicament parent), respectivement .
Absorption cutanée chez le lapin
Dans une étude de pharmacocinétique cutanée chez le lapin, l'absorption cutanée du dutastéride dans CAPMUL (oléate de glycéryle) chez le lapin a entraîné des concentrations sériques de 2,7 à 40,5 mcg/h/mL pour des doses de 1 à 20 mg/mL, respectivement, ou 56 % à 100 % de dutastéride appliqué à absorber dans des conditions occluses et prolongées. Les capsules de gélatine molle AVODART administrées par voie orale contiennent 0,5 mg de dutastéride dissous dans un mélange de mono-di-glycérides d'acide caprylique/caprique et de butylhydroxytoluène. Le dutastéride dans l'eau a été très peu absorbé chez le lapin (2 000 mg/kg).
Etudes cliniques
Monothérapie
AVODART 0,5 mg/jour (n = 2 167) ou un placebo (n = 2 158) a été évalué chez des hommes atteints d'HBP dans trois essais multicentriques, contrôlés par placebo, en double aveugle, d'une durée de 2 ans, chacun avec des prolongations ouvertes de 2 ans ( n = 2 340). Plus de 90 % de la population de l'essai était blanche. Les sujets étaient âgés d'au moins 50 ans avec un PSA sérique ≥ 1,5 ng/mL et
Effet sur les scores de symptômes
Les symptômes ont été quantifiés à l'aide de l'AUA-SI, un questionnaire qui évalue les symptômes urinaires (vidange incomplète, fréquence, intermittence, urgence, flux faible, effort et nycturie) en les évaluant sur une échelle de 0 à 5 pour un score total possible de 35, avec scores de symptômes totaux numériques plus élevés représentant une plus grande sévérité des symptômes. Le score AUA-SI initial dans les 3 essais était d'environ 17 unités dans les deux groupes de traitement.
Les sujets recevant du dutastéride ont obtenu une amélioration statistiquement significative des symptômes par rapport au placebo au mois 3 dans 1 essai et au mois 12 dans les 2 autres essais pivots. Au mois 12, la diminution moyenne par rapport au départ des scores totaux des symptômes AUA-SI dans les 3 essais regroupés était de -3,3 unités pour le dutastéride et de -2,0 unités pour le placebo avec une différence moyenne entre les 2 groupes de traitement de -1,3 (intervalle : -1,1 à -1,5 unités dans chacun des 3 essais, P
Ces essais ont été conçus de manière prospective pour évaluer les effets sur les symptômes en fonction de la taille de la prostate au départ. Chez les hommes avec des volumes prostatiques ≥ 40 cc, la diminution moyenne était de -3,8 unités pour le dutastéride et de -1,6 unités pour le placebo, avec une différence moyenne entre les 2 groupes de traitement de -2,2 au mois 24. Chez les hommes avec des volumes prostatiques
Figure 1 : Changement du score AUA-SIa par rapport au départ (essais randomisés, en double aveugle, contrôlés par placebo regroupés)
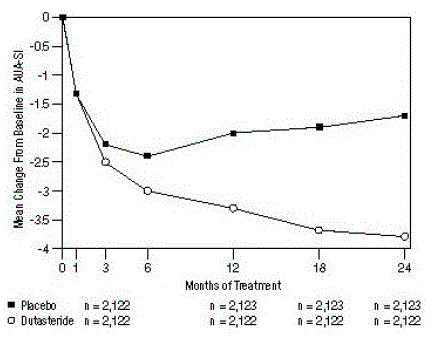
un score AUA-SI allant de 0 à 35.
Effet sur la rétention urinaire aiguë et la nécessité d'une chirurgie liée à l'HBP
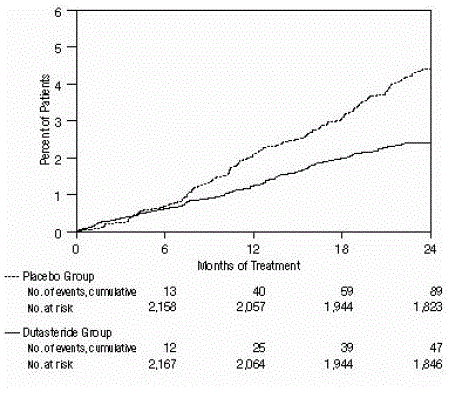
L'efficacité a également été évaluée après 2 ans de traitement par l'incidence des RAU nécessitant un cathétérisme et une intervention chirurgicale urologique liée à l'HBP. Par rapport au placebo, AVODART a été associé à une incidence statistiquement significativement plus faible de RAU (1,8 % pour AVODART versus 4,2 % pour le placebo, P
Figure 2 : Pourcentage de sujets développant une rétention urinaire aiguë sur une période de 24 mois (essais randomisés, en double aveugle, contrôlés par placebo regroupés)
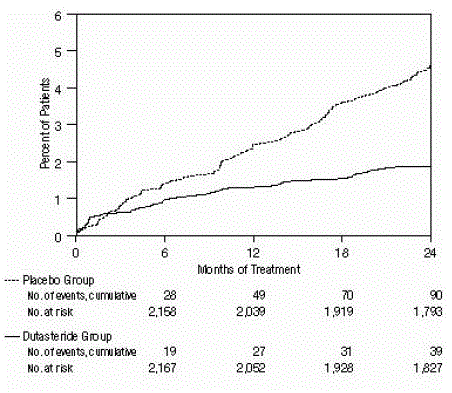
Figure 3 : Pourcentage de sujets opérés pour une hyperplasie bénigne de la prostate sur une période de 24 mois (essais randomisés, en double aveugle, contrôlés par placebo regroupés)
Effet sur le volume de la prostate
Un volume prostatique d'au moins 30 cc mesuré par échographie transrectale était requis pour participer à l'essai. Le volume moyen de la prostate à l'entrée dans l'essai était d'environ 54 cc.
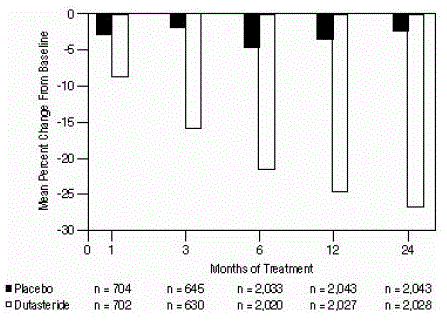
Des différences statistiquement significatives (AVODART 0,5 mg versus placebo) ont été notées lors de la première mesure du volume de la prostate post-traitement dans chaque essai (mois 1, mois 3 ou mois 6) et se sont poursuivies jusqu'au mois 24. Au mois 12, la variation moyenne en pourcentage du le volume de la prostate dans les 3 essais regroupés était de -24,7 % pour le dutastéride et de -3,4 % pour le placebo ; la différence moyenne (dutastéride moins placebo) était de -21,3 % (intervalle : -21,0 % à -21,6 % dans chacun des 3 essais, P
Figure 4 : Variation en pourcentage du volume de la prostate par rapport au départ (essais randomisés, en double aveugle, contrôlés par placebo regroupés)
Effet sur le débit urinaire maximal
Un débit urinaire de pointe moyen (Qmax) ≤ 15 mL/s était requis pour participer à l'essai. Qmax était d'environ 10 mL/sec au départ dans les 3 essais pivots.
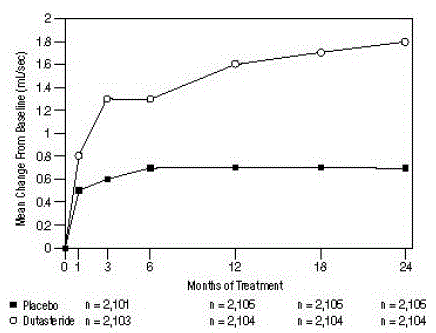
Les différences entre les 2 groupes étaient statistiquement significatives par rapport à l'inclusion au mois 3 dans les 3 essais et se sont maintenues jusqu'au mois 12. Au mois 12, l'augmentation moyenne du Qmax dans les 3 essais regroupés était de 1,6 ml/s pour AVODART 0,5 mg et 0,7 ml /sec pour le placebo ; la différence moyenne (dutastéride moins placebo) était de 0,8 mL/sec (plage : 0,7 à 1,0 mL/sec dans chacun des 3 essais, P
Figure 5 : Changement de Qmax par rapport au départ (essais randomisés, en double aveugle, contrôlés par placebo regroupés)
Résumé des essais cliniques
Les données de 3 grands essais d'efficacité bien contrôlés démontrent que le traitement par AVODART (0,5 mg une fois par jour) réduit le risque d'intervention chirurgicale liée à la fois à la RAU et à l'HBP par rapport au placebo, améliore les symptômes liés à l'HBP, diminue le volume de la prostate et augmente le débits urinaires. Ces données suggèrent qu'AVODART 0,5 mg arrête le processus pathologique de l'HBP chez les hommes présentant une hypertrophie de la prostate.
Combinaison avec la thérapie alpha-bloquante (CombAT)
L'efficacité de l'association (AVODART 0,5 mg/jour plus tamsulosine 0,4 mg/jour, n = 1 610) a été comparée à AVODART 0,5 mg seul (n = 1 623) ou à la tamsulosine seule (n = 1 611) dans une étude multicentrique de 4 ans, randomisée , essai en double aveugle. Les critères d'admission aux essais étaient similaires à ceux des essais d'efficacité en monothérapie en double aveugle contrôlés par placebo décrits à la section 14.1. Quatre-vingt-huit pour cent (88 %) de la population participant à l'essai étaient de race blanche. Environ 52 % des sujets avaient déjà été exposés à un traitement inhibiteur de la 5 alpha-réductase ou antagoniste alpha-adrénergique. Sur les 4 844 sujets randomisés pour recevoir le traitement, 69 % des sujets du groupe association, 67 % du groupe recevant AVODART 0,5 mg et 61 % du groupe tamsulosine ont suivi 4 ans de traitement en double aveugle.
Effet sur le score des symptômes
Les symptômes ont été quantifiés à l'aide des 7 premières questions de l'International Prostate Symptom Score (IPSS) (identique à l'AUA-SI). Le score initial était d'environ 16,4 unités pour chaque groupe de traitement. La thérapie combinée était statistiquement supérieure à chacun des traitements en monothérapie en ce qui concerne la diminution du score des symptômes au mois 24, le point temporel principal pour ce critère d'évaluation. Au mois 24, les changements moyens par rapport à l'inclusion (±ET) des scores IPSS totaux des symptômes étaient de -6,2 (±7,14) pour l'association, -4,9 (±6,81) pour AVODART et -4,3 (±7,01) pour la tamsulosine, avec une différence moyenne entre l'association et AVODART 0,5mg de -1,3 unités (P
Figure 6 : Changement du score international des symptômes de la prostate par rapport au départ sur une période de 48 mois (essai randomisé, en double aveugle, en groupes parallèles [essai CombAT])
![International Prostate Symptom Score Change from Baseline over a 48Month Period (Randomized, Double-blind, Parallel-Group Trial [CombAT Trial]) - Illustration](/pics/c284b3823673fb3d3d6ede12e72e38a3.jpg)
Effet sur la rétention urinaire aiguë ou la nécessité d'une chirurgie liée à l'HBP
Après 4 ans de traitement, la thérapie combinée avec AVODART 0,5 mg et la tamsulosine n'a pas apporté d'avantage par rapport à la monothérapie avec AVODART pour réduire l'incidence de la RAU ou de la chirurgie liée à l'HBP.
Effet sur le débit urinaire maximal
Le Qmax initial était d'environ 10,7 mL/sec pour chaque groupe de traitement. La thérapie combinée était statistiquement supérieure à chacun des traitements en monothérapie en ce qui concerne l'augmentation du Qmax au mois 24, le point temporel principal pour ce critère d'évaluation. Au mois 24, les augmentations moyennes du Qmax par rapport au départ (±ET) étaient de 2,4 (±5,26) mL/sec pour l'association, 1,9 (±5,10) mL/sec pour AVODART 0,5 mg et 0,9 (±4,57) mL/sec pour tamsulosine, avec une différence moyenne entre l'association et AVODART de 0,5 mL/sec (P = 0,003 ; [IC 95 % : 0,17 ; 0,84]), et entre l'association et la tamsulosine de 1,5 mL/sec (P
L'amélioration supplémentaire du Qmax du traitement combiné par rapport à la monothérapie avec AVODART 0,5 mg n'était plus statistiquement significative au mois 48.
Figure 7 : Changement de Qmax par rapport à la ligne de base sur une période de 24 mois (essai randomisé, en double aveugle, en groupes parallèles [essai CombAT])
![Qmax Change from Baseline over a 24-Month Period (Randomized, Double-blind, Parallel-Group Trial [CombAT Trial]) - Illustration](/pics/23e887a40ba8ee4acf0740dcb7d18364.jpg)
Effet sur le volume de la prostate
Le volume moyen de la prostate à l'entrée dans l'essai était d'environ 55 cc. Au mois 24, le point temporel principal pour ce critère d'évaluation, les changements moyens en pourcentage par rapport au départ (±ET) du volume de la prostate étaient de -26,9 % (±22,57) pour le traitement combiné, de -28,0 % (±24,88) pour AVODART 0,5 mg et 0% (±31,14) pour la tamsulosine, avec une différence moyenne entre l'association et AVODART 0,5mg de 1,1% (P = NS ; [IC 95% : -0,6 ; 2,8]), et entre l'association et la tamsulosine de -26,9% (P
INFORMATIONS PATIENTS
AVODART (av o dart) (dutastéride) gélules
AVODART 0,5 mg est réservé aux hommes.
Lisez ces informations patient avant de commencer à prendre AVODART 0,5 mg et chaque fois que vous recevez une recharge. Il peut y avoir de nouvelles informations. Ces informations ne remplacent pas une discussion avec votre fournisseur de soins de santé au sujet de votre état de santé ou de votre traitement.
Qu'est-ce qu'AVODART 0,5 mg ?
AVODART 0,5 mg est un médicament délivré sur ordonnance qui contient du dutastéride. AVODART 0,5 mg est utilisé pour traiter les symptômes de l'hyperplasie bénigne de la prostate (HBP) chez les hommes présentant une hypertrophie de la prostate pour :
- améliorer les symptômes,
- réduire le risque de rétention urinaire aiguë (blocage complet du flux urinaire),
- réduire le risque de nécessiter une intervention chirurgicale liée à l'HBP.
Qui ne devrait PAS prendre AVODART ?
Ne prenez pas AVODART si vous êtes :
- enceinte ou susceptible de le devenir. AVODART 0,5 mg peut nuire à votre bébé à naître. Les femmes enceintes ne doivent pas toucher les capsules AVODART. Si une femme enceinte d'un bébé de sexe masculin reçoit suffisamment d'AVODART 0,5 mg dans son corps en avalant ou en touchant AVODART, le bébé de sexe masculin peut naître avec des organes sexuels anormaux. Si une femme enceinte ou une femme en âge de procréer entre en contact avec des gélules AVODART 0,5 mg qui fuient, la zone de contact doit être immédiatement lavée à l'eau et au savon.
- un enfant ou un adolescent.
- allergique au dutastéride ou à l'un des ingrédients d'AVODART. Voir la fin de cette notice pour la liste complète des ingrédients d'AVODART.
- allergique à d'autres inhibiteurs de la 5 alpha-réductase, par exemple les comprimés PROSCAR (finastéride) ®.
Que dois-je dire à mon professionnel de la santé avant de prendre AVODART 0,5 mg ?
Avant de prendre AVODART 0,5 mg, informez votre professionnel de la santé si vous :
- avoir des problèmes de foie
Informez votre fournisseur de soins de santé de tous les médicaments que vous prenez, y compris les médicaments sur ordonnance et en vente libre, les vitamines et les suppléments à base de plantes. AVODART et d'autres médicaments peuvent interagir les uns avec les autres, entraînant des effets secondaires. AVODART 0,5 mg peut affecter le fonctionnement d'autres médicaments, et d'autres médicaments peuvent affecter le fonctionnement d'AVODART 0,5 mg.
Connaissez les médicaments que vous prenez. Conservez-en une liste pour la montrer à votre fournisseur de soins de santé et à votre pharmacien lorsque vous recevez un nouveau médicament.
Comment dois-je prendre AVODART ?
- Prendre 1 gélule AVODART une fois par jour.
- Avalez les gélules d'AVODART 0,5 mg entières. Ne pas écraser, mâcher ou ouvrir les gélules d'AVODART 0,5 mg car le contenu de la gélule peut irriter les lèvres, la bouche ou la gorge.
- Vous pouvez prendre AVODART 0,5 mg avec ou sans nourriture.
- Si vous oubliez une dose, vous pouvez la prendre plus tard dans la journée. Ne rattrapez pas la dose oubliée en prenant 2 doses le lendemain.
Que dois-je éviter pendant que je prends AVODART 0,5 mg ?
- Vous ne devez pas donner de sang pendant que vous prenez AVODART ou pendant les 6 mois suivant l'arrêt d'AVODART. Ceci est important pour empêcher les femmes enceintes de recevoir AVODART par transfusion sanguine.
Quels sont les effets secondaires possibles d'AVODART ?
AVODART peut provoquer des effets secondaires graves, notamment :
- Réactions allergiques rares et graves, notamment :
- gonflement du visage, de la langue ou de la gorge
- réactions cutanées graves, telles que desquamation de la peau
Consultez immédiatement un médecin si vous avez ces réactions allergiques graves.
- Risque accru de développer une forme plus grave de cancer de la prostate.
Les effets secondaires les plus courants d'AVODART comprennent :
- difficulté à obtenir ou à maintenir une érection (impuissance)*
- une diminution de la libido (libido)*
- problèmes d'éjaculation*
- seins hypertrophiés ou douloureux. Si vous remarquez des grosseurs dans les seins ou un écoulement du mamelon, vous devriez en parler à votre fournisseur de soins de santé.
*Certains de ces événements peuvent persister après l'arrêt de la prise d'AVODART.
Une humeur dépressive a été rapportée chez des patients recevant AVODART.
Il a été démontré qu'AVODART 0,5 mg réduit le nombre de spermatozoïdes, le volume de sperme et le mouvement des spermatozoïdes. Cependant, l'effet d'AVODART 0,5 mg sur la fertilité masculine n'est pas connu.
Test d'antigène spécifique de la prostate (PSA) : Votre fournisseur de soins de santé peut vérifier si vous avez d'autres problèmes de prostate, y compris le cancer de la prostate, avant de commencer et pendant que vous prenez AVODART. Un test sanguin appelé PSA (antigène spécifique de la prostate) est parfois utilisé pour voir si vous pourriez avoir un cancer de la prostate. AVODART réduira la quantité de PSA mesurée dans votre sang. Votre fournisseur de soins de santé est conscient de cet effet et peut toujours utiliser le PSA pour voir si vous pourriez avoir un cancer de la prostate. Les augmentations de vos niveaux de PSA pendant le traitement par AVODART (même si les niveaux de PSA sont dans la plage normale) doivent être évaluées par votre professionnel de la santé.
Dites à votre fournisseur de soins de santé si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas.
Ce ne sont pas tous les effets secondaires possibles avec AVODART. Pour plus d'informations, demandez à votre fournisseur de soins de santé ou à votre pharmacien.
Appelez votre médecin pour obtenir des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
Comment dois-je conserver AVODART 0,5 mg ?
- Conservez les capsules AVODART 0,5 mg à température ambiante (59 °F à 86 °F ou 15 °C à 30 °C).
- Les gélules d'AVODART 0,5 mg peuvent se déformer et/ou se décolorer si elles sont conservées à des températures élevées.
- N'utilisez pas AVODART 0,5 mg si vos gélules sont déformées, décolorées ou fuient.
- Jetez en toute sécurité les médicaments dont vous n'avez plus besoin.
Gardez AVODART 0,5 mg et tous les médicaments hors de la portée des enfants.
Les médicaments sont parfois prescrits à des fins autres que celles énumérées dans une notice patient. N'utilisez pas AVODART 0,5 mg pour une affection pour laquelle il n'a pas été prescrit. Ne donnez pas AVODART à d'autres personnes, même si elles présentent les mêmes symptômes que vous. Cela peut leur nuire.
Cette notice d'information destinée aux patients résume les informations les plus importantes sur AVODART. Si vous souhaitez plus d'informations, parlez-en à votre fournisseur de soins de santé. Vous pouvez demander à votre pharmacien ou à votre professionnel de la santé des informations sur AVODART 0,5 mg destinées aux professionnels de la santé.
Pour plus d'informations, rendez-vous sur www.AVODART.com ou appelez le 1-888-825-5249.
Quels sont les ingrédients d'AVODART 0,5 mg ?
Ingrédient actif: dutastéride.
Ingrédients inactifs : butylhydroxytoluène, oxyde de fer (jaune), gélatine (provenant de sources bovines certifiées exemptes d'ESB), glycérine, mono-diglycérides d'acide caprylique/caprique, dioxyde de titane et encre rouge comestible.
Comment fonctionne AVODART ?
La croissance de la prostate est causée par une hormone dans le sang appelée dihydrotestostérone (DHT). AVODART réduit la production de DHT dans le corps, entraînant un rétrécissement de l'hypertrophie de la prostate chez la plupart des hommes. Bien que certains hommes aient moins de problèmes et de symptômes après 3 mois de traitement par AVODART, une période de traitement d'au moins 6 mois est généralement nécessaire pour voir si AVODART 0,5 mg fonctionnera pour vous.
Ces informations destinées aux patients ont été approuvées par la Food and Drug Administration des États-Unis.